
よくある質問
よくあるご質問については
以下のページにまとめております
・トラブルシューティングガイド
・解析ソフトダウンロード
・納品レポートの見方



1. QC全般について
 HoldとFail判定の違いを教えてください。
HoldとFail判定の違いを教えてください。
 QCに使用する液量を教えてください。
QCに使用する液量を教えてください。
 もっとたくさん液量を送ったはずなのですが。
もっとたくさん液量を送ったはずなのですが。
 サンプルを希釈している理由を教えてください。
サンプルを希釈している理由を教えてください。
 サンプルを再送したいのですが、費用は発生しますか。
サンプルを再送したいのですが、費用は発生しますか。
 再送の期限はありますか。
再送の期限はありますか。
 再送したいのですが、初回サンプルも捨てないでください。
再送したいのですが、初回サンプルも捨てないでください。
 再送サンプルのQC結果次第では初回サンプルを使用して進めてください。
再送サンプルのQC結果次第では初回サンプルを使用して進めてください。
 再送の方法を教えてください。
再送の方法を教えてください。
 一度サンプルを返送いただいてから再調整、サンプル再送を行いたいです。
一度サンプルを返送いただいてから再調整、サンプル再送を行いたいです。
 Failサンプルだけでなく、Passサンプルも再送したいです。
Failサンプルだけでなく、Passサンプルも再送したいです。
 QC結果が悪かったサンプルの代わりに、同じサンプルではなく別のサンプルを送付したいです。
QC結果が悪かったサンプルの代わりに、同じサンプルではなく別のサンプルを送付したいです。
 1サンプルに対し同時に2サンプルを送付・QC結果を確認後、
1サンプルに対し同時に2サンプルを送付・QC結果を確認後、
シーケンスを行うサンプルを選択したいです。
 初回サンプルと再送サンプルを混合して進めたいです。
初回サンプルと再送サンプルを混合して進めたいです。
 事前に測定した濃度との違いが大きいです。
事前に測定した濃度との違いが大きいです。
 事前にQCをせずにサンプルを送ってもいいですか。
事前にQCをせずにサンプルを送ってもいいですか。
 微量サンプルのため、サンプルQCは省略し、
微量サンプルのため、サンプルQCは省略し、
全量を使用してライブラリ作製をお願いしたいです。
 ライブラリ調製時のinput DNA / RNA量を教えてください。
ライブラリ調製時のinput DNA / RNA量を教えてください。
 DNA / RNA 量が基準値未満ですが、このまま進めたいです。
DNA / RNA 量が基準値未満ですが、このまま進めたいです。
 一部のサンプルのみ進行し、その結果を確認してから残りのサンプルの進行を決めたいです。
一部のサンプルのみ進行し、その結果を確認してから残りのサンプルの進行を決めたいです。
 お勧めの抽出キットはありますか。
お勧めの抽出キットはありますか。
2.RNA QC について
 RIN (RNA Integrity Number) とはなんでしょうか。
RIN (RNA Integrity Number) とはなんでしょうか。
 rRNA ratio (%) とはなんでしょうか。
rRNA ratio (%) とはなんでしょうか。
 RINとrRNA ratioが0.1となっています。事前測定ではもっと高かったのですが。
RINとrRNA ratioが0.1となっています。事前測定ではもっと高かったのですが。
 DV200 (%) とはなんでしょうか。
DV200 (%) とはなんでしょうか。
 5s peak(small peak)とはなんでしょうか。
5s peak(small peak)とはなんでしょうか。
 DNA混入の疑いと判定された理由はなんでしょうか。
DNA混入の疑いと判定された理由はなんでしょうか。
またこのまま進めた場合のリスクを教えてください。
 分解が進んでいますが、このまま進めた場合のリスクを教えてください。
分解が進んでいますが、このまま進めた場合のリスクを教えてください。
 分解が進んでいますが、キットを変更して進めることはできますか。
分解が進んでいますが、キットを変更して進めることはできますか。
 分解が進んでいる理由として何が考えられますか。
分解が進んでいる理由として何が考えられますか。
 凍結させる組織の大きさはRNAの質に影響しますか。
凍結させる組織の大きさはRNAの質に影響しますか。
3.DNA / PCR産物QC について
 DNA精製をしていただけますか。
DNA精製をしていただけますか。
 DIN (DNA Integrity Number) とはなんでしょうか。
DIN (DNA Integrity Number) とはなんでしょうか。
 DIN値が低かったですが、再抽出時、気を付ける点があれば教えてください。
DIN値が低かったですが、再抽出時、気を付ける点があれば教えてください。
 PCR増幅がされないのはなぜでしょうか。
PCR増幅がされないのはなぜでしょうか。
4. Library QC について
 Library QCの内容を教えてください。
Library QCの内容を教えてください。
 事前に測定したサイズとQC結果のサイズに乖離があります。
事前に測定したサイズとQC結果のサイズに乖離があります。
 Multi peak (broad peak, high molecular Peak)とのことですが、
Multi peak (broad peak, high molecular Peak)とのことですが、
このまま進めても問題ないですか。
 small peak (adapter dimer) とはどのピークのことですか。
small peak (adapter dimer) とはどのピークのことですか。
また結果にどのような影響がありますか。
 濃度が基準値未満ですが、シーケンスは可能ですか。
濃度が基準値未満ですが、シーケンスは可能ですか。
 濃度が低いので追加で送るサンプルと混合して進めていただけますか。
濃度が低いので追加で送るサンプルと混合して進めていただけますか。
 濃縮してシーケンスへ進めていただけますか。
濃縮してシーケンスへ進めていただけますか。
 精製を行っていただけますか。
精製を行っていただけますか。
 Poolingはどのように行いますか。
Poolingはどのように行いますか。
 ライブラリQC結果の報告が送られてきません。
ライブラリQC結果の報告が送られてきません。
5. BioChipQC について
 濃縮は可能ですか。
濃縮は可能ですか。
 RNA混入疑いとのことですが、RNase処理をお願いできますか。
RNA混入疑いとのことですが、RNase処理をお願いできますか。
 純度が低いですが、精製をお願いできますか。
純度が低いですが、精製をお願いできますか。
6. Cancer Panel QC について
 純度が低いですが、精製をお願いできますか。
純度が低いですが、精製をお願いできますか。
 再度サンプルを調整したのですが(FFPEからのゲノム抽出)、
再度サンプルを調整したのですが(FFPEからのゲノム抽出)、
純度が2.0以上と高く出てしまいます。何か対応策はありますか。


1.納品結果md5sum値について
md5sum値とはなんですか。
md5sum値が一致しません。
md5sum値の確認方法について教えて下さい。
2. 納品結果fastqfileについて
fastqfileとはなんですか。
fastqfileを使用するソフトウェアについて教えて下さい。
fastq.gz fileの解凍方法について教えて下さい。
fastqfileをダウンロードしましたがサイズが小さいです。
fastq.gz fileをダウンロードしていますが終わりません。
fastqfileのダウンロードができません。(Not Foundと表示される)
ダウンロード期限を過ぎてしまったがダウンロードしたいです。
アダプター配列について教えて下さい。
3. 納品結果HDD納品について
パスワードの案内メールがきていないです。
パスワードを入力したがエラーになります。
パスワードを入力したが解除が完了しません。
4. 納品結果解析物について
どのファイルをダウンロードすればいいですか。
解析結果を見たがファイルが見れません。
解析結果内のhtmlレポートの画像が表示されません。
Raw dataが2個ずつあるのはなぜですか。
vcfファイルの確認方法について教えて下さい。
PDFレポートが複数あるがダウンロードするファイルは片方のみで大丈夫ですか。
- ヒトWGS解析
- ヒトWES解析
- RNA-Seq解析
- ASV解析
※本”納品レポートの見方”中のデータは一般的な納品例となっております。
お手元のレポートと一致しない場合もございますので、ご了承ください。
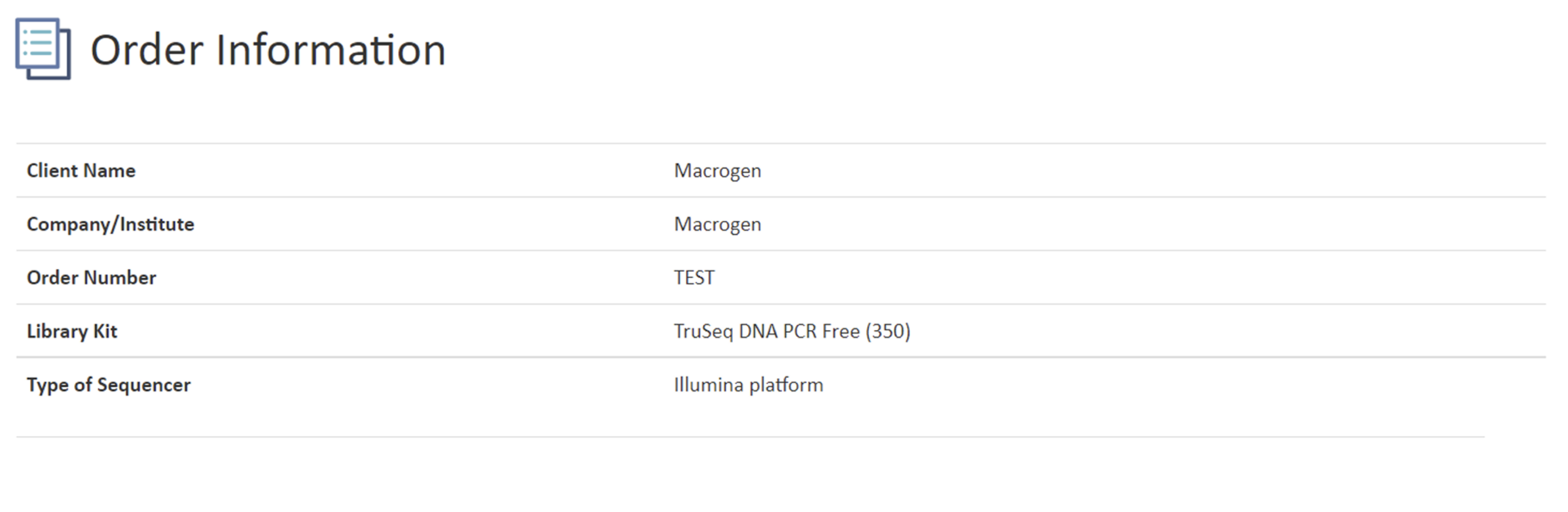
ORDER INFORMATION
こちらの項目では、本案件で使用しましたライブラリ調製キット、シーケンサー機種をご案内しています。
シーケンサー機種名につきまして、下記例では”illumina platform”との記載になっていますが、
データ量保証のプランでWGS解析をご依頼の場合、NovaSeq 6000 および NovaSeq X plus シーケンサーを使用しての対応となります。
illumiina NovaSeq6000 Sequencing System
https://jp.illumina.com/systems/sequencing-platforms/novaseq.html
illumiina NovaSeq X plus Sequencing System
https://jp.illumina.com/systems/sequencing-platforms/novaseq-x-plus.html
DELIVERABLES
Raw dataおよび解析結果のダウンロードリンクが記載されています。
Raw data(fastqファイル)は論文投稿時に行うデータベース登録時に、また、弊社でのデータ保管期間経過後に、追加データ解析をご希望の場合に必要なファイルです。
ダウンロードできる期間は約2週間となっておりますので、必ず期間中に全てのファイルをダウンロードして下さい。また、ダウンロードしたfastq.gzファイルはファイルに破損がないかの確認のため、必ず”md5sum値”の照合を行ってください。ご確認をお願いしております。
確認、照合方法につきましては”CAUTION”をご確認ください。HDD納品対応の場合には、HDD内容について詳細記載があります。
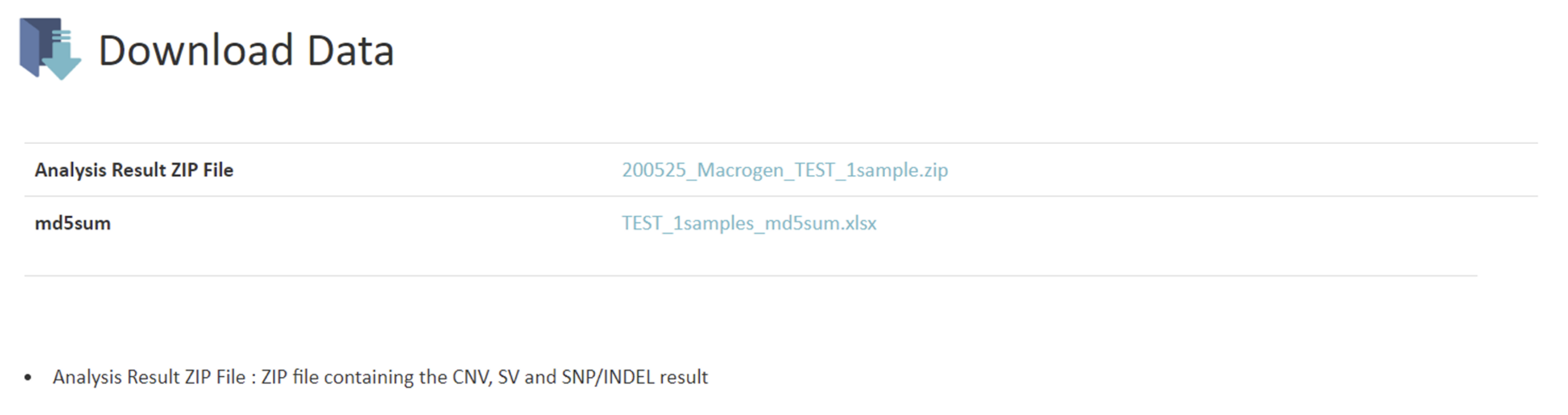
ANALYSIS RESULT
例1
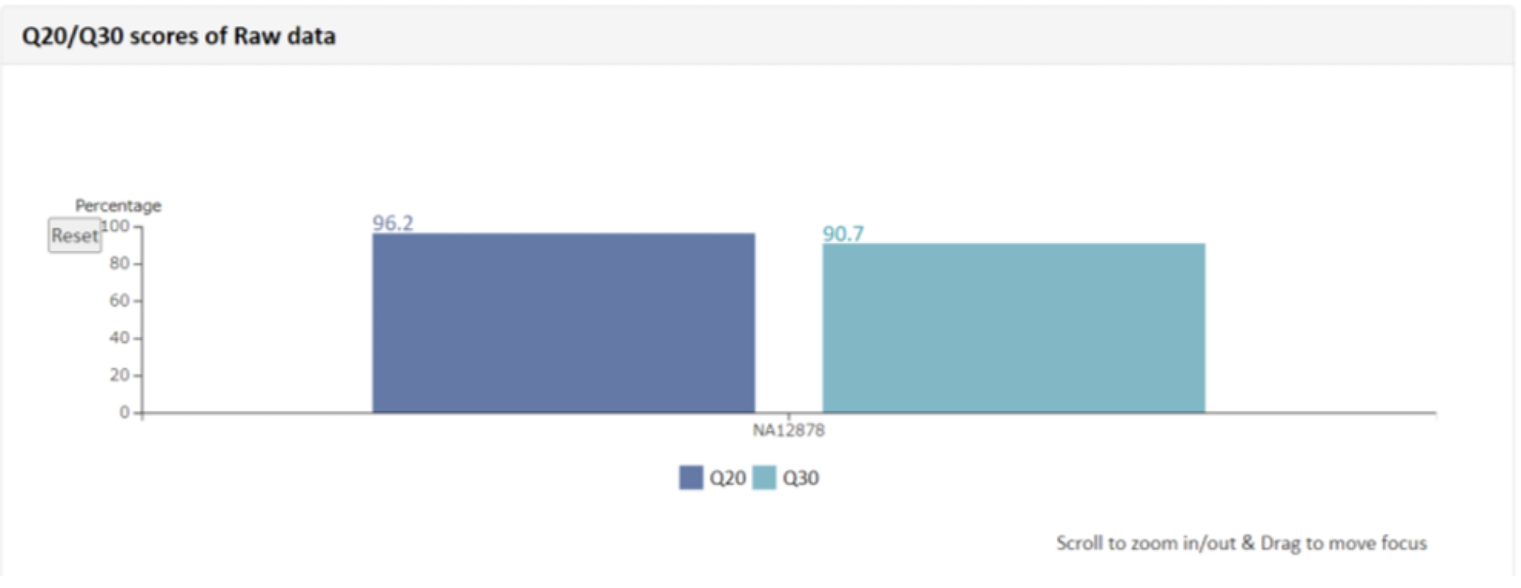
こちらの項目では得られたリードデータのリード数、GC%、Qualityの確認結果を記載しています。例1 Q20/Q30 scores of Raw data・Phredというプログラムで算出したQuality Score(QS) = Phredクオリティスコアベースコールにおけるエラー率の予測指標。Q20: PhredQSが20以上の塩基の割合Q30: PhredQSが30以上の塩基の割合※QSの詳細はResult File Descriptionにも記載があります。
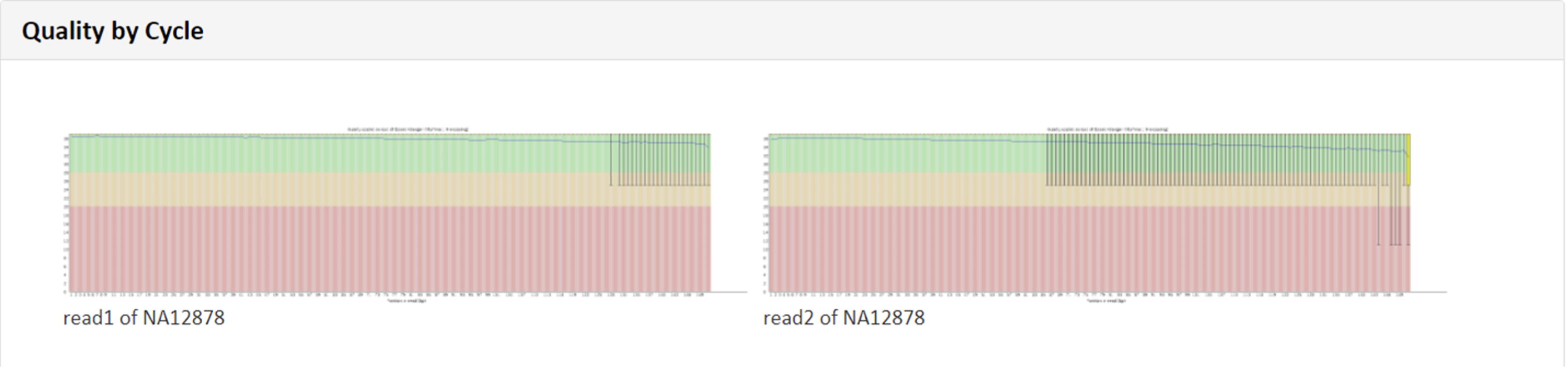
例2 Quality by Cycle
FASTQCというプログラムで算出したQSを基に、Forward(read1)およびReverse(read2)について、リードの位置ごとのQSを図示しています。・縦軸:QS、横軸:リード上での位置、緑色領域:Good Quality、黄色領域:Acceptable Quality、赤色領域:Bad Quality を示しており、得られたリードを平均して評価した際に、どのQualityにあたるのか確認できます。
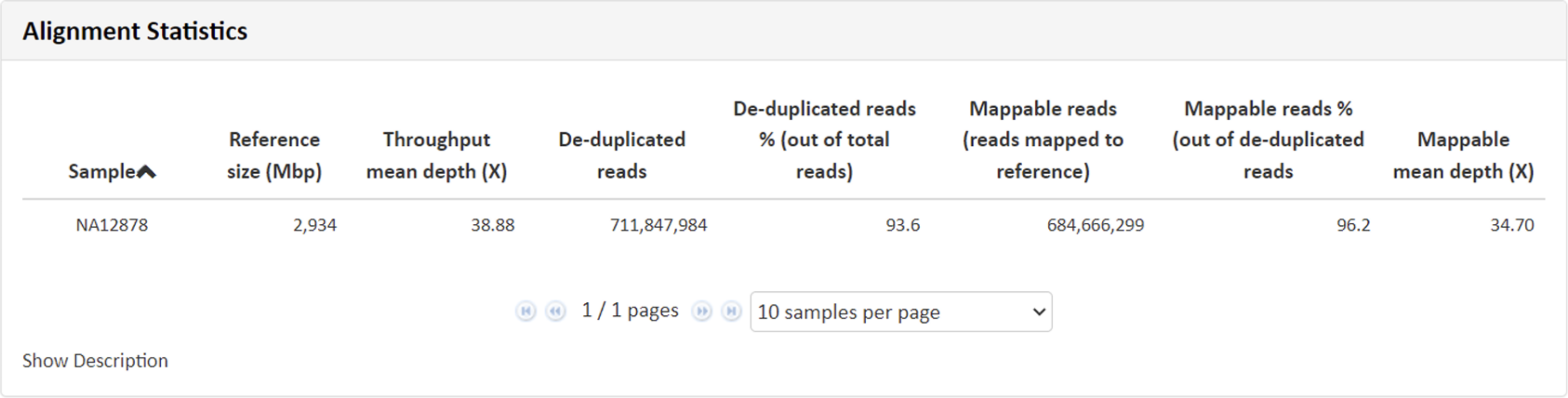
例1 Alignment Statistics
こちらの項目では得られたリードデータをリファレンス情報にマッピングし、マッピングの状況から算出致しました、depth、リード数、カバー率、インサートサイズなどのデータを記載しています。
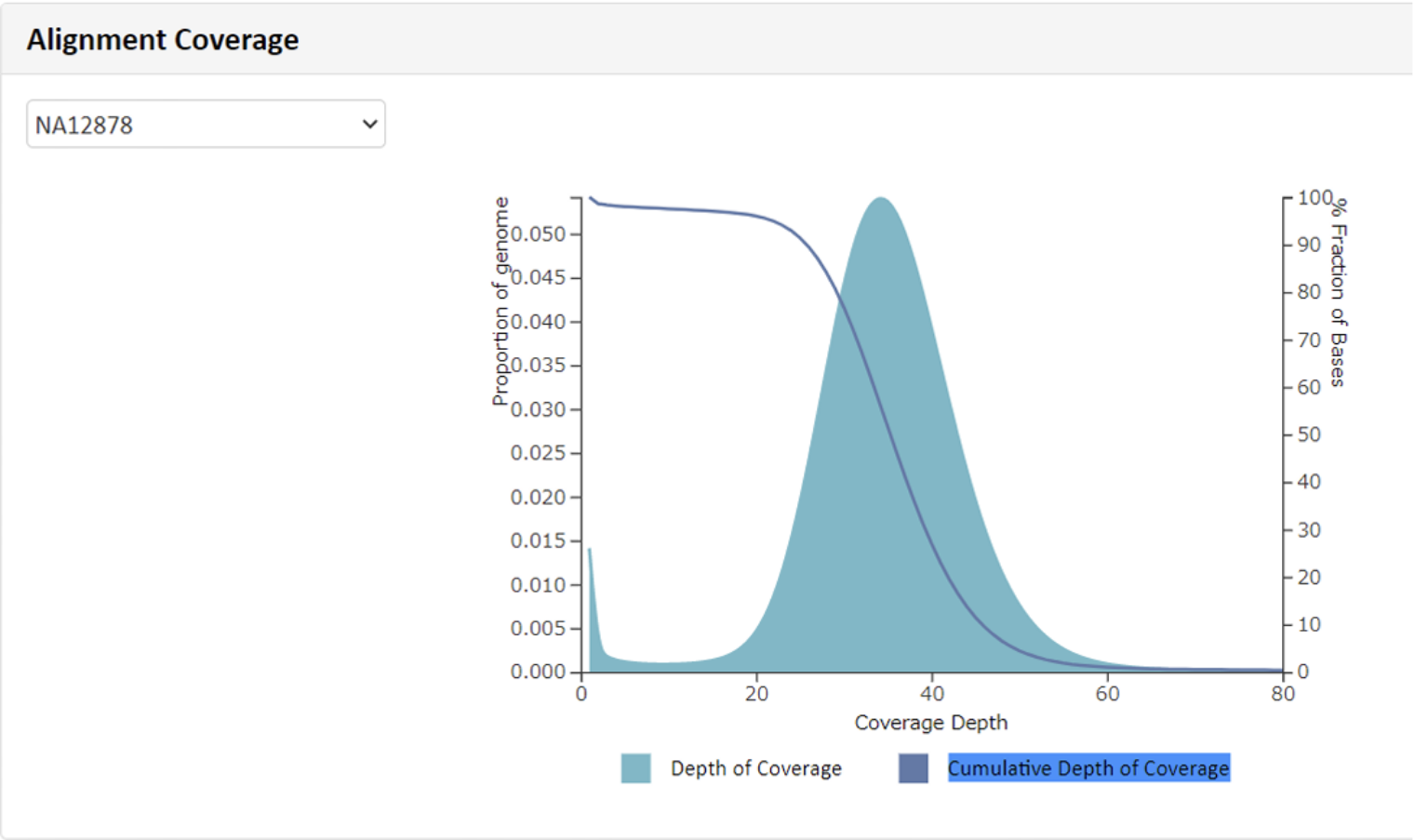
例2 Alignment Coverage
Alignment解析により算出した領域ごとのdepthおよび累積深度分布を図示しています。累積深度とは、ある深さ以上の対象領域が占める割合のことであり、Depth:40手前より割合が低下していますので、例1mean depth:38.88と一致した形状となっています。“Show Description”をクリックできる場合、その部分をクリックすると、数値および名称につきまして詳細と凡例を確認できます。
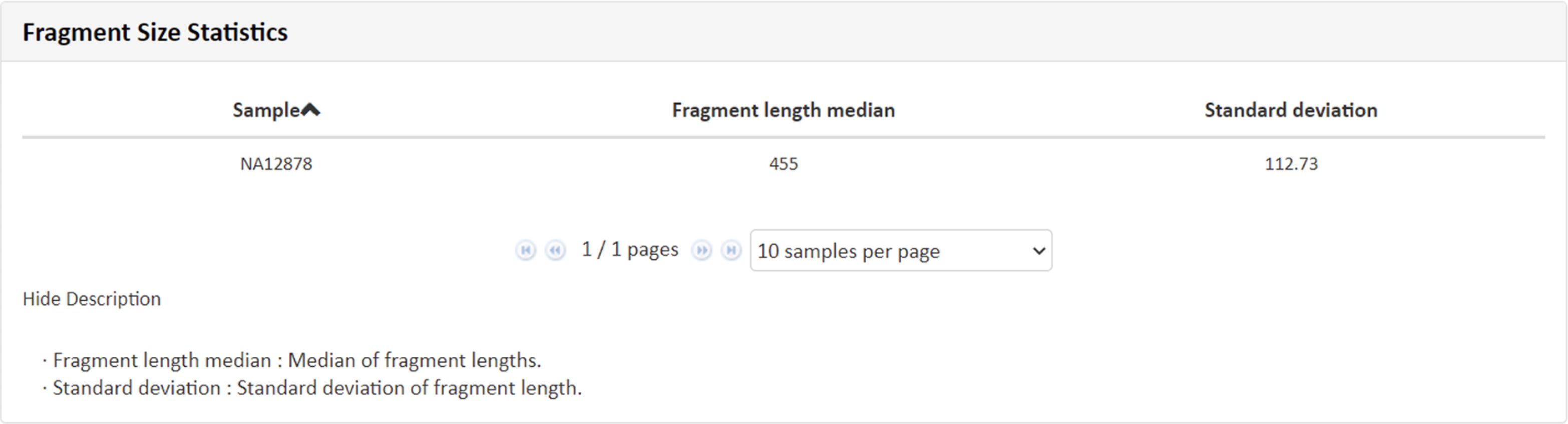
例3 Fragment Size Statistics

例4 Fragment Size Histogram
Mapping解析によりリードがマッピングされた領域を”Fragment”と定義し、Fragmentsizeおよび累積サイズ頻度の分布を図示しています。累積サイズ頻度とは、ある長さ以上の対象領域が占める割合であり、Insert size:350前後より割合が大きく低下しており、例3Fragment length 平均値(455)からStandard deviation(112)を引いた値(343)と一致した形状となっています。“Show Description”をクリックできる場合、その部分をクリックすると、数値および名称につきまして詳細と凡例を確認できます。
こちらの項目では得られたリードデータをリファレンス情報にマッピングし、マッピングの状況から算出致しました、一塩基多型(SNP)、InDelなどのデータを記載しています。
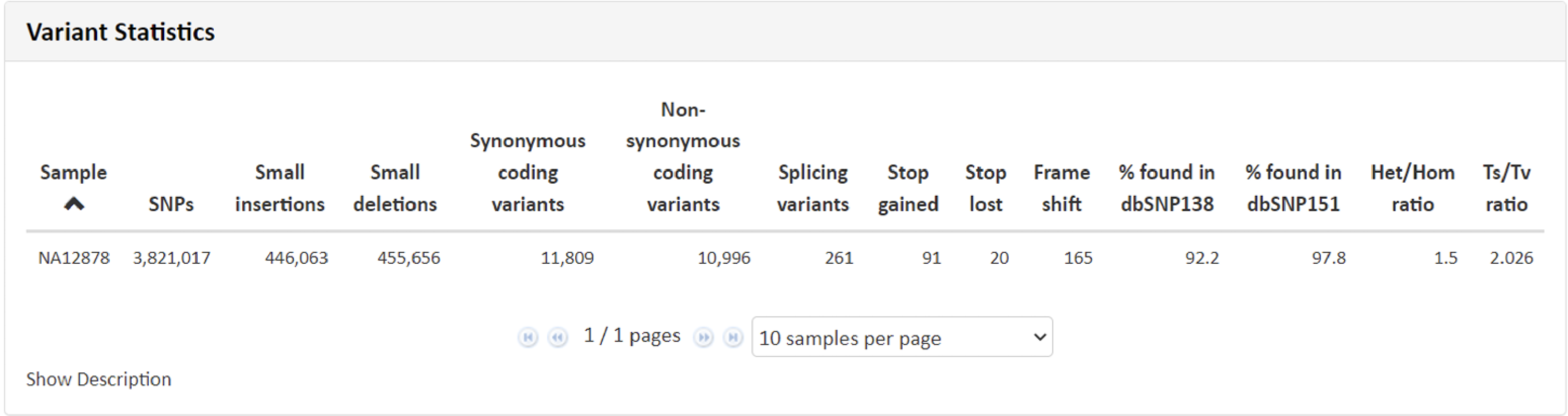
例1 Variant Statistics
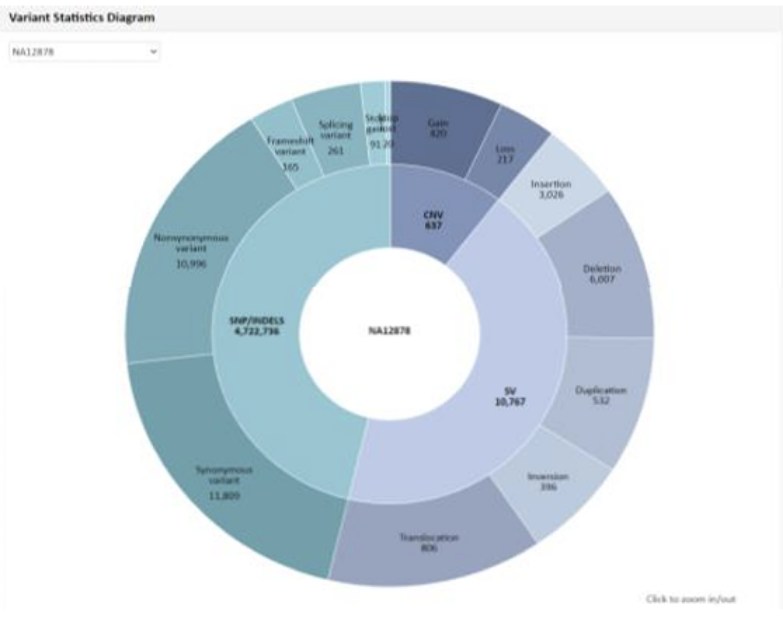
例2 Variant Statistics Diagram
Mapping解析により検出したSNP、InDel、構造多型(SV)、コピー数多型(CNV)の、総数および内訳分類を円グラフで表示しています。
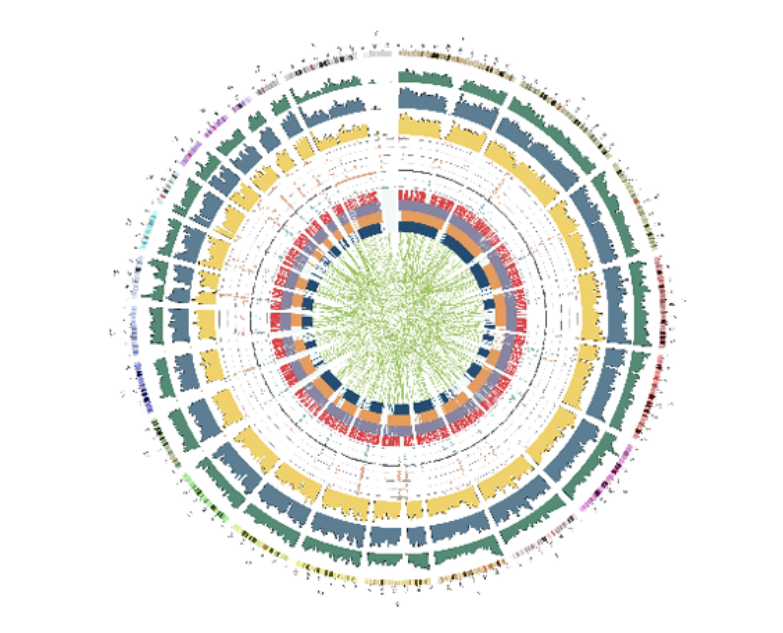
例3 Circos
ヒトサンプルの場合は、Circos plotを作成しています。染色体ごとのSNP、InDel、SV、CNVの局在と関連を確認できます。“Show Description”をクリックできる場合、その部分をクリックすると、数値および名称につきまして詳細と凡例を確認できます。
DESCRIPTION
こちらの項目では、ライブラリ作製からシーケンス解析までを”Experimental”、得られたリードデータを基に実施したデータ解析を”Analysis”と分類し、各々のワークフローを簡単に記載しています。

例1 Experimental Overview
こちらの項目では、ライブラリ作製からシーケンス解析までを”Experimental”、得られたリードデータを基に実施したデータ解析を”Analysis”と分類し、各々のワークフローを簡単に記載しています。
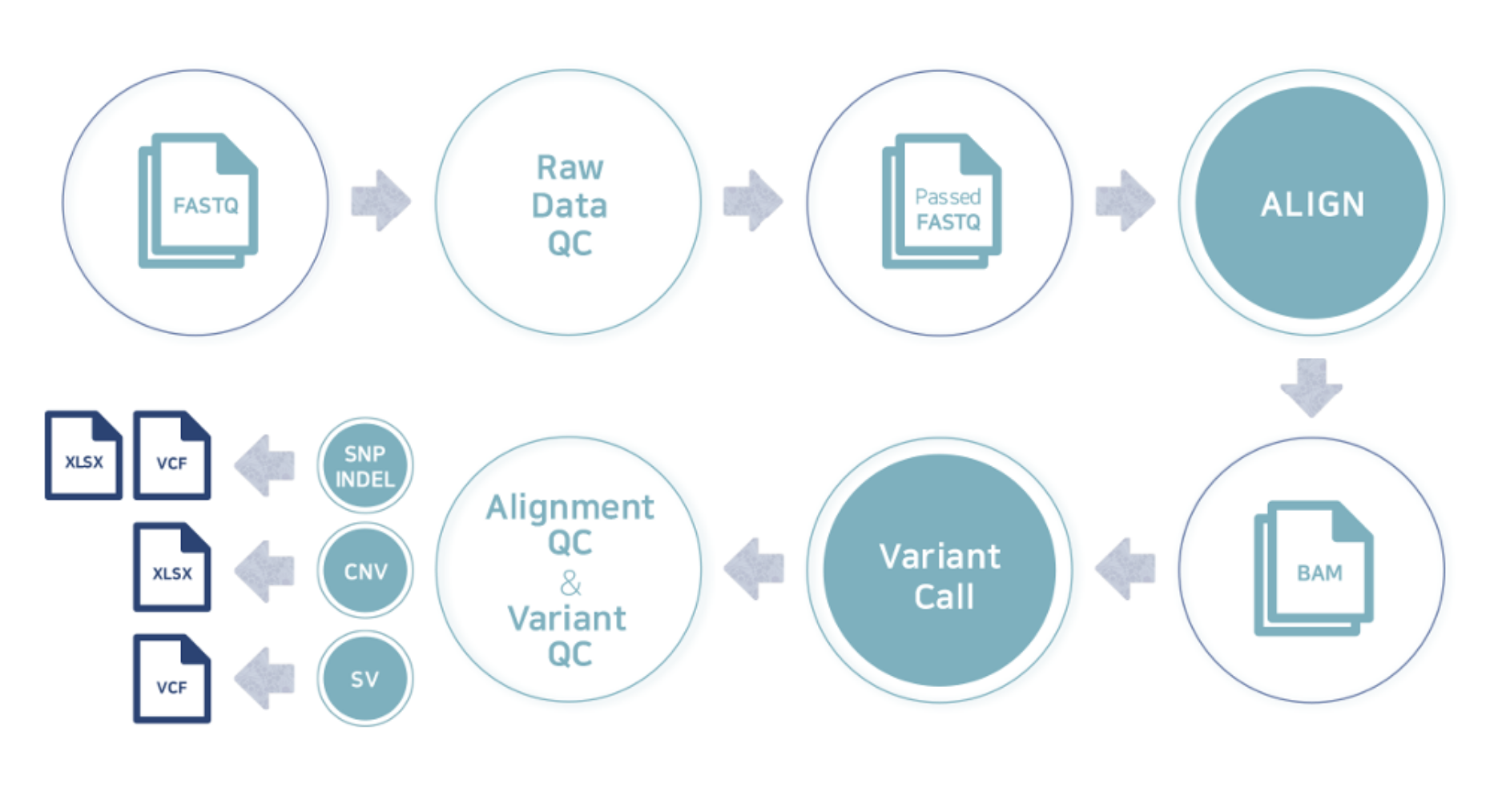
例2 Whole genome resequencing analysis process
こちらの項目では、データ解析に使用しておりますプログラムおよびデータベースの、バージョン、パラメータなど詳細内容を記載しています。論文記載、Method作成の際にご参照ください。③Analysis Tools ④AnaysisDatabase本解析における用語の定義については後述の、“AnaysisDatabase”項目にて英文で詳細をまとめています。
例1 Effect (Sequence Ontology)

”Column Description”として、SNP/InDel解析結果
Excel file (XXXX(Sample名)_chrX(染色体番号).xlsx)の各カラムについての詳細を記載しています。
- CHROM:染色体番号・POS:位置
- REF:リファレンス上の塩基情報
- ALT:サンプルで変異が確認された塩基情報
- REF_DP:リファレンス情報の塩基のアレル深度
- ALT_DP:サンプルで確認された変異塩基のアレル深度
- QUAL:REF/ALT多型が存在する確率をPhredでスケーリングした値。Phredスケール:-10*log(1-p)となります。こちらのスコアが10の場合:10分の1の確率でエラーがあることを示し、100の場合:10^10分の1の確率であることを示します。
- MQ:Mapping Quality
- Zygosity:Homo/Hetero
-
FILTER:下記条件でフィルター設定し、条件分けしています
(例2)。該当の位置ですべてのフィルターを通過した場合はPASS記載となります。フィルタに合格していない場合、失敗したフィルタのコードがセミコロンで区切られた形式で記載となります。

- Effect:Sequence Ontologyの用語を用いたアノテーション結果。複数の結果の場合、‘&’で連結して表記しています。
- Putative_Impact:推定される影響度/削除度。”HIGH, MODERATE, LOW, MODIFIER”として簡易推定結果を記載しています。
- Gene_Name:一般的な遺伝子名(HUGO Gene Nomenclature Committee:HGNC基準)。遺伝子間に位置する変異の場合、最も近い遺伝子を記載しています。
- Feature_Type:転写物、モチーフ、miRNAなど特定領域の特徴を記載しています。
- Feature_ID:Transcript ID、Motif ID、miRNA、ChipSeqピーク、Histone markなど、ID登録されている場合、そのIDを記載しています。
- Transcript_BioType:ENSEMBL biotypes、Coding、Noncodingについて記載しています。
- Rank/Total:エクソンまたはイントロンのランク/エクソンまたはイントロンの総数。
- HGVS.c:HGVS表記による変異(DNAレベル)
- HGVS.p:HGVS表記による変異(アミノ酸レベル)
上記以外の項目につきましては、”Column Description”をご確認ください。
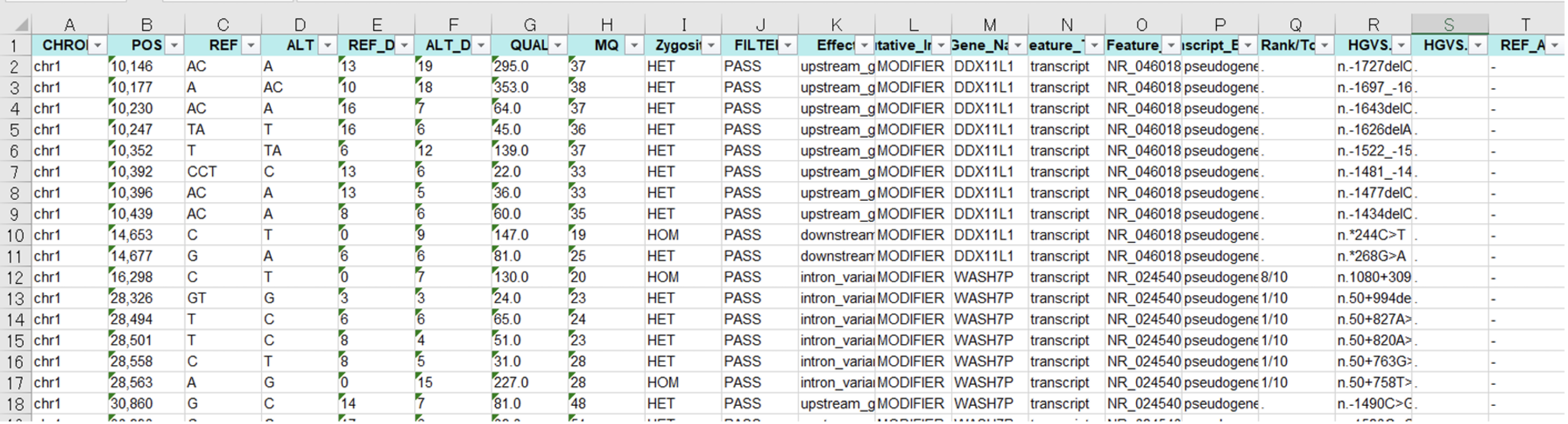
例3 SNP/InDel解析結果Excel file
こちらの項目では、納品しておりますFileの種類・形式につきまして詳細を記載しています。
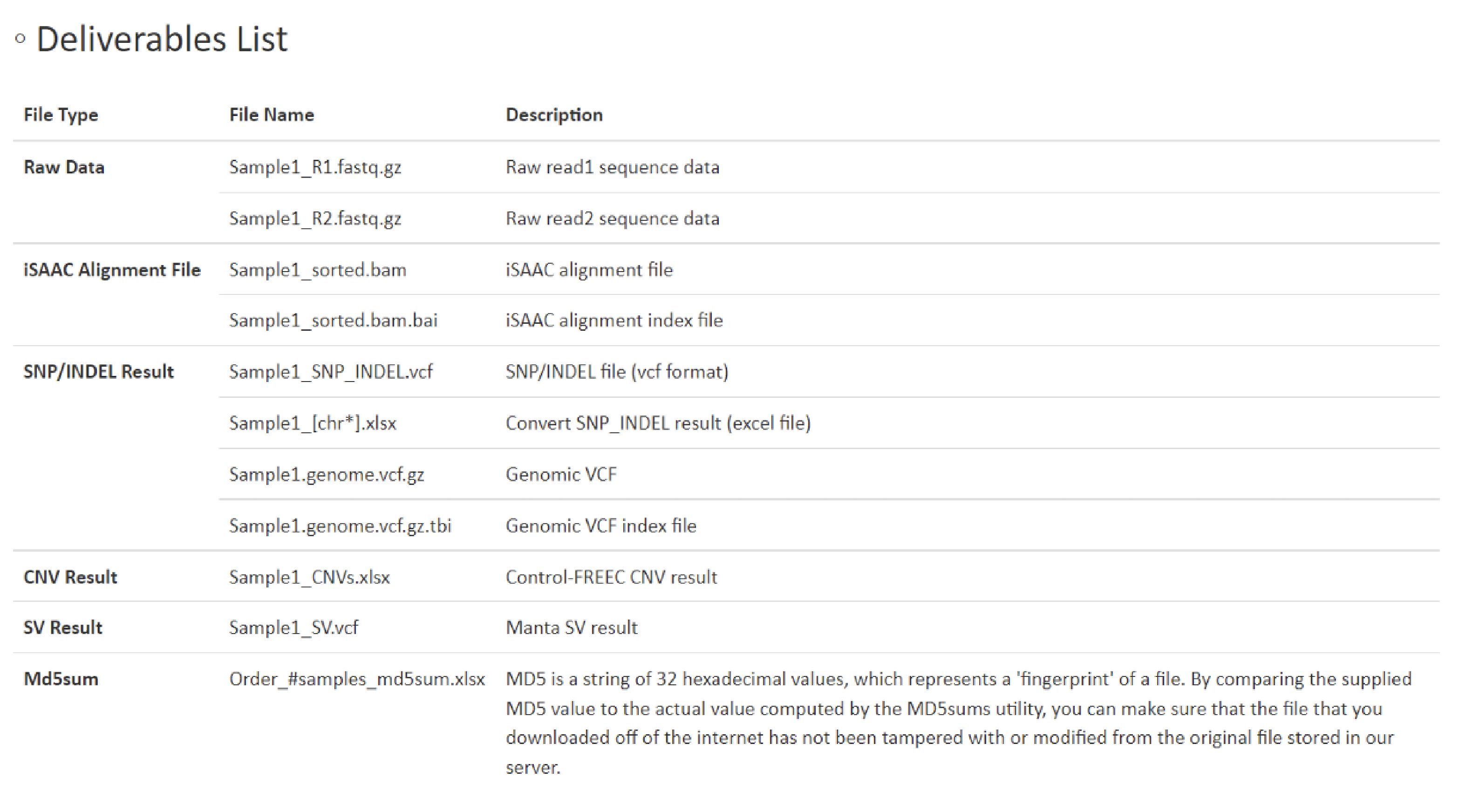
また、ヒトサンプルを解析した際に納品している、CNV解析結果Excel file (XXXX(Sample名)_CNVs.xlsx) SV解析結果vcf file (XXXX(Sample名)_SV.vcf)の各カラムについての詳細もこちらに記載しています。上記file を確認される際には、こちらの記載をご参照ください。
ダウンロード納品のご依頼の際は弊社html形式のレポートサイトの、
DELIVERABLESの頁にRaw dataおよび解析結果のダウンロードリンクがあります。
Raw dataはデータベース登録時や、弊社でのデータ保管期間経過後に、追加データ解析をご希望の場合に必要なファイルとなります。
ダウンロードできる期間は約2週間となっておりますので、必ず全てのファイルをダウンロード頂きますよう、宜しくお願い致します。
なお、2週間経過後も1ヵ月はデータを保管しておりますので、再度ダウンロードが必要な場合は
ngs@macrogen-japan.co.jpまでご連絡ください。
ダウンロードしたFastq.gzファイルもしくはHDD内のFastq.gzファイルは、ファイルの解凍前に”md5sum値”のご確認くだい。
”QuickHash-GUI”という、フリーのアプリケーションもございます。https://www.quickhash-gui.org/downloads/
もしお手持ちのソフトウェアが無ければこちらをご取得下さい。
QuickHash-GUIでのmd5sum値の確認方法は下記となります。
“QuickHash-GUI.exe”アプリケーションを起動します(例1)。
①“FileS”タブをクリックし、
②“Algorithm”、”MD5”を選択してください。md5sum値を確認したいfastqファイルが入っているフォルダを
③“Select Directory” から選択していただきますと、自動的に解析が進行致します。
出力は、csvファイルまたはtxtファイルとして保存することができます。
使用するシステムの性能により、処理に時間がかかる場合がございます。表示された数字と、レポートに記載のmd5sum値の一致を確認できましたら作業完了となります。あわせてQuickHash-GUIのユーザーマニュアルもご確認ください(例2)。
例1 QuickHash-GUI起動画面

例2 QuickHash-GUIfile中身

※本”納品レポートの見方”中のデータは一般的な納品例となっております。
お手元のレポートと一致しない場合もございますので、ご了承ください。
ORDER INFORMATION
こちらの項目では、本案件で使用しましたライブラリ調製キット、シーケンサー機種をご案内しています。シーケンサー機種名につきまして、下記例では”illumine platform”との記載になっていますが、データ量保証のプランでWES解析をご依頼の場合、NovaSeq6000 および NovaSeq X plus シーケンサーを使用しての対応となります。
illumiina NovaSeq6000 Sequencing System
https://jp.illumina.com/systems/sequencing-platforms/novaseq.html
illumiina NovaSeq X plus Sequencing System
https://jp.illumina.com/systems/sequencing-platforms/novaseq-x-plus.html

DELIVERABLES
Raw dataおよび解析結果のダウンロードリンクが記載されています。
Raw data(fastqファイル)は論文投稿時に行うデータベース登録時に、また、弊社でのデータ保管期間経過後に、追加データ解析をご希望の場合に必要なファイルです。
ダウンロードできる期間は約2週間となっておりますので、必ず期間中に全てのファイルをダウンロードして下さい。また、ダウンロードしたfastq.gzファイルはファイルに破損がないかの確認のため、必ず”md5sum値”の照合を行ってください。ご確認をお願いしております。
確認、照合方法につきましては“CAUTION”をご確認ください。HDD納品対応の場合には、HDD内容について詳細記載があります。
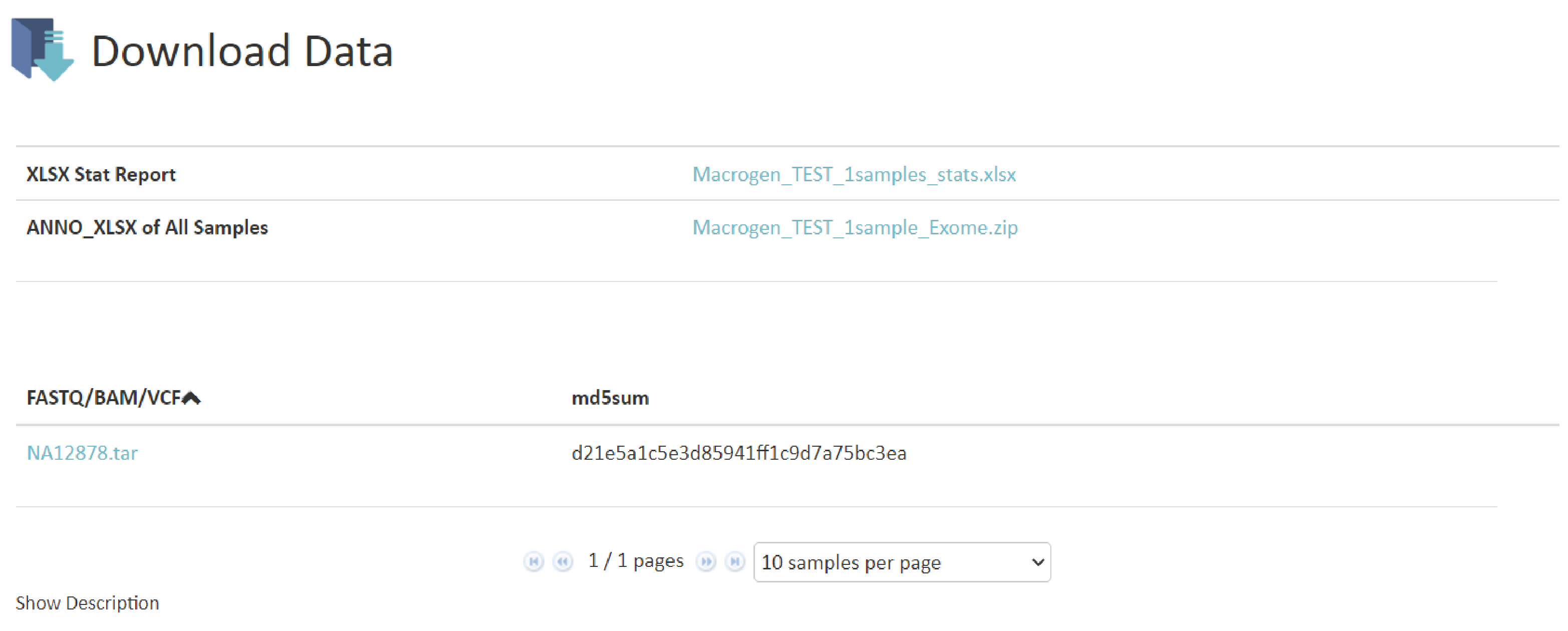
ANALYSIS RESULT

例1
こちらの項目では得られたリードデータのリード数、GC%、Qualityの確認結果を記載しています。例1 Q20/Q30 scores of Raw data・Phredというプログラムで算出したQuality Score(QS) = Phredクオリティスコアベースコールにおけるエラー率の予測指標。Q20: PhredQSが20以上の塩基の割合Q30: PhredQSが30以上の塩基の割合※QSの詳細はResult File Descriptionにも記載があります。
例2 Quality by Cycle
FASTQCというプログラムで算出したQSを基に、Forward(read1)およびReverse(read2)について、リードの位置ごとのQSを図示しています。・縦軸:QS、横軸:リード上での位置、緑色領域:Good Quality、黄色領域:Acceptable Quality、赤色領域:Bad Quality を示しており、得られたリードを平均して評価した際に、どのQualityにあたるのか確認できます。
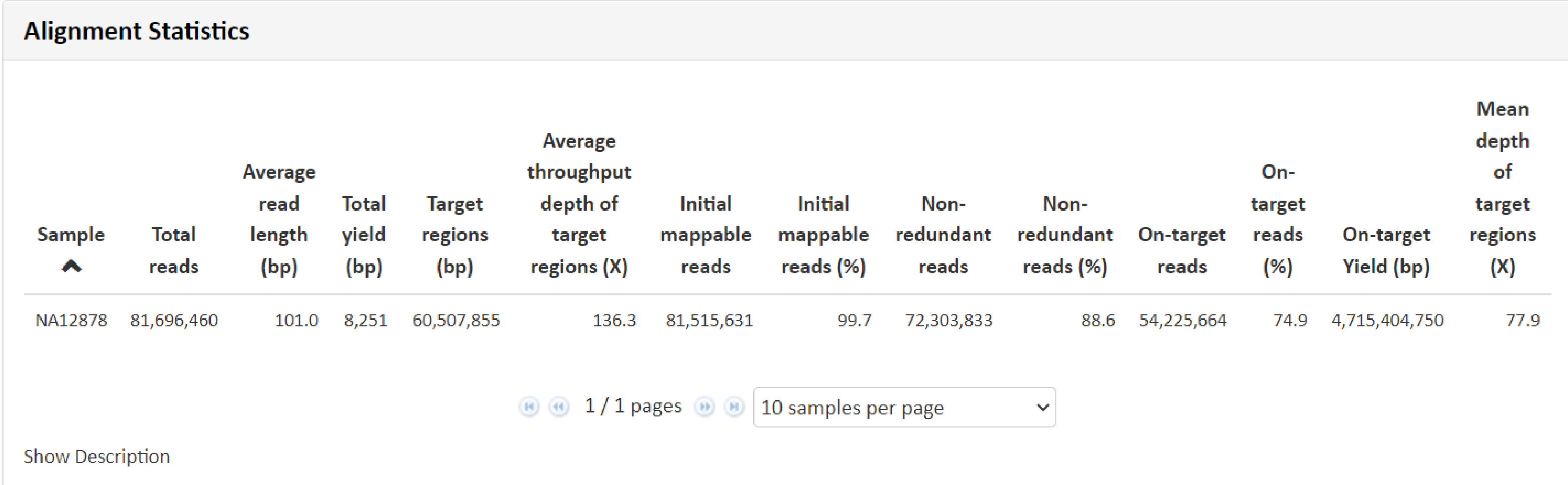
例1 Alignment Statistics
こちらの項目では得られたリードデータをリファレンス情報にマッピングし、マッピングの状況から算出致しました、depth 、 リード数、カバー率、リード長、一定数(1x, 10x, 20x, 30x, 以上のカバー率の領域の割合などのデータを記載しています。

例2 Alignment Coverage
Alignment解析により算出した領域ごとの depthおよび累積深度分布を図示しています。累積深度とは、ある深さ以上の対象領域が占める割合のことであり、Depth:80手前より 80% 以下と、割合が大きく低下していますので、例1 mean depth 77.9 と一致した形状となっています。“Show Description” をクリックできる場合、その部分をクリックすると、数値および名称につきまして詳細と凡例を確認できます。
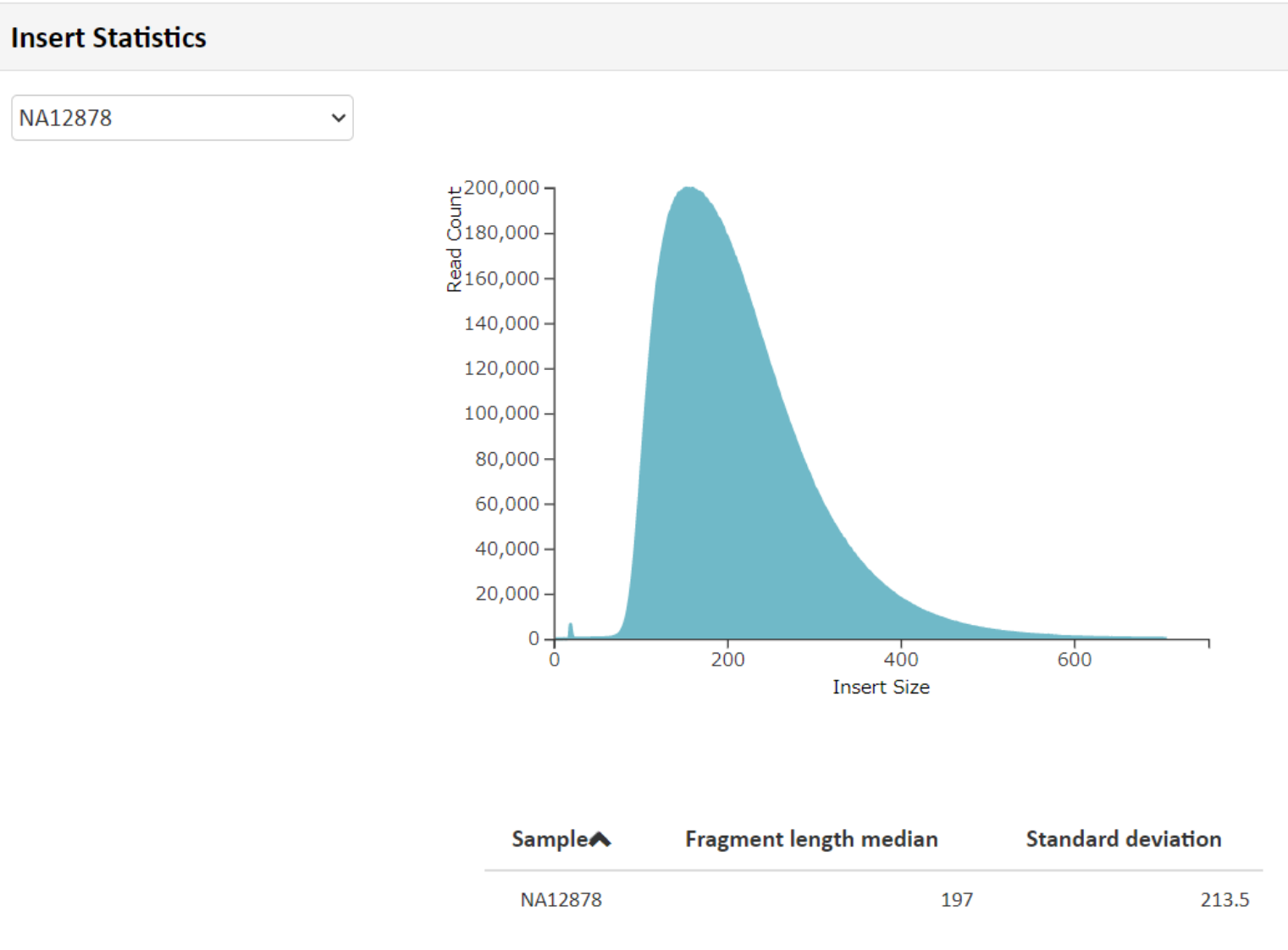
例3 Insert StatisticsMapping
解析によりリードがマッピングされた領域を ” と定義し、Fragmentsize の分布を図示しております。Fragment平均長: 197 と算出され、波形データのピークの位置と一致しています。
こちらの項目では得られたリードデータをリファレンス情報にマッピングし、マッピングの状況から算出致しました、マッピングの状況から算出致しました。一塩基多型(SNP) 、 InDel 、ヘテロ/ホモの割合などのデータを記載しています。
例1 Variant Statistics
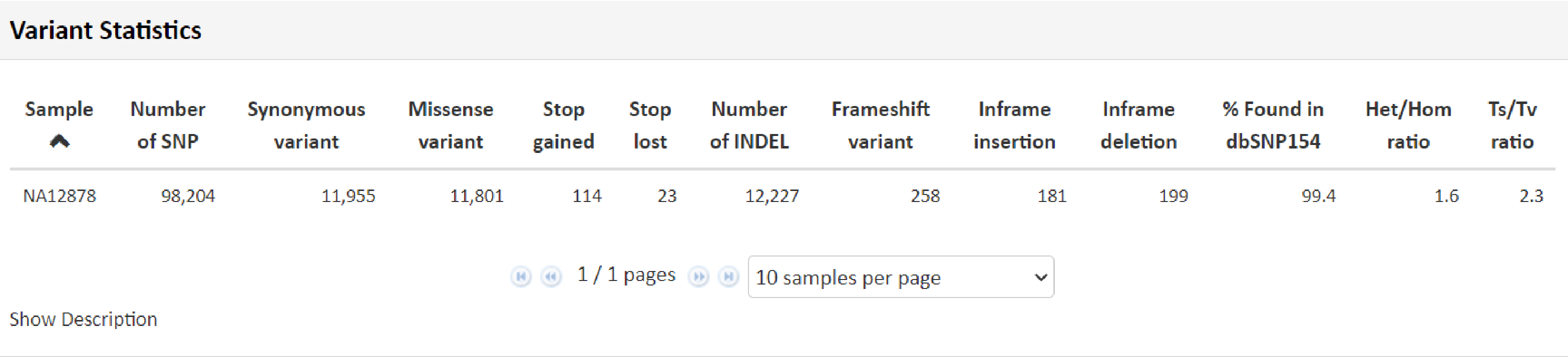
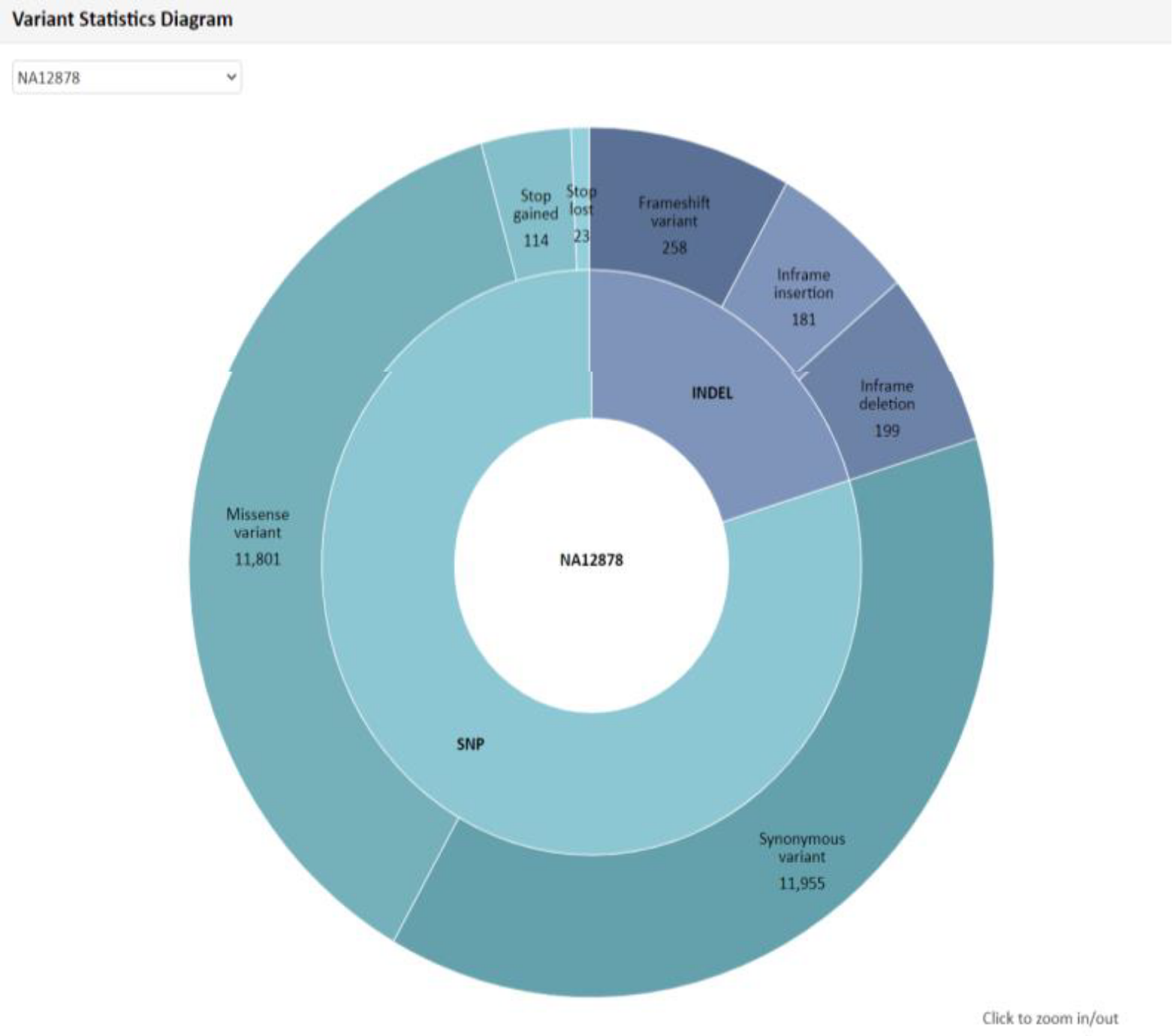
例2 Variant Statistics Diagram Mapping解析により検出した SNP 、 InDel につきまして、総数および内訳分類を円グラフで表示しています。“Show Description” をクリックできる場合、その部分をクリックすると、数値および名称につきまして詳細と凡例を確認できます。
DESCRIPTION
こちらの項目では、ライブラリ作製からシーケンス解析までを”Experimental”、得られたリードデータを基に実施したデータ解析を”Analysis”と分類し、各々のワークフローを簡単に記載しています。
例1 Experimental Overview
こちらの項目では、ライブラリ作製からシーケンス解析までを”Experimental”、得られたリードデータを基に実施したデータ解析を”Analysis”と分類し、各々のワークフローを簡単に記載しています。
例2 Whole genome resequencing analysis process
こちらの項目では、データ解析に使用しておりますプログラムおよびデータベースの、バージョン、パラメータなど詳細内容を記載しています。論文記載、Method作成の際にご参照ください。③Analysis Tools ④AnaysisDatabase本解析における用語の定義については後述の、“AnaysisDatabase”項目にて英文で詳細をまとめています。
例1 Effect (Sequence Ontology)
”Annotation Column”として、SNP/InDel解析結果
Excel file (XXXX(Sample名)_chrX(染色体番号).xlsx)の各カラムについての詳細を記載しています。
- CHROM:染色体番号
- POS:位置
- REF:リファレンス上の塩基情報
- ALT:サンプルで変異が確認された塩基情報
- DP:変異があったアレル全体のリード数
- AD:変異があったリード数
※Allele Frequency(AF)の数値は ”AD/DP* で求めることができます。 - QUAL:REF/ALT多型が存在する確率をPhredでスケーリングした値。Phredスケール:-10*log(1-p)となります。こちらのスコアが10の場合:10分の1の確率でエラーがあることを示し、100の場合:10^10分の1の確率であることを示します。
- MQ:Mapping Quality
- Zygosity:Homo/Hetero
-
FILTER:下記条件でフィルター設定し、条件分けしています
(例2)。該当の位置ですべてのフィルターを通過した場合はPASS記載となります。フィルタに合格していない場合、失敗したフィルタのコードがセミコロンで区切られた形式で記載となります。

例2 フィルター設定の詳細
- Effect:Sequence Ontologyの用語を用いたアノテーション結果。複数の結果の場合、‘&’で連結して表記しています。
- Putative_Impact:推定される影響度/削除度。”HIGH, MODERATE, LOW, MODIFIER”として簡易推定結果を記載しています。
- Gene_Name:一般的な遺伝子名(HUGO Gene Nomenclature Committee:HGNC基準)。遺伝子間に位置する変異の場合、最も近い遺伝子を記載しています。
- Feature_Type:転写物、モチーフ、miRNAなど特定領域の特徴を記載しています。
- Feature_ID:Transcript ID、Motif ID、miRNA、ChipSeqピーク、Histone markなど、ID登録されている場合、そのIDを記載しています。
- Transcript_BioType:ENSEMBL biotypes、Coding、Noncodingについて記載しています。
- Rank/Total:エクソンまたはイントロンのランク/エクソンまたはイントロンの総数。
- HGVS.c:HGVS表記による変異(DNAレベル)
- HGVS.p:HGVS表記による変異(アミノ酸レベル)
上記以外の項目につきましては、結果レポートのAppendix>”Annotation Column”をご確認ください。

例3 SNP/InDel解析結果Excel file
こちらの項目では、納品しておりますFileの種類・形式につきまして詳細を記載しています。
例1 Deliverables List
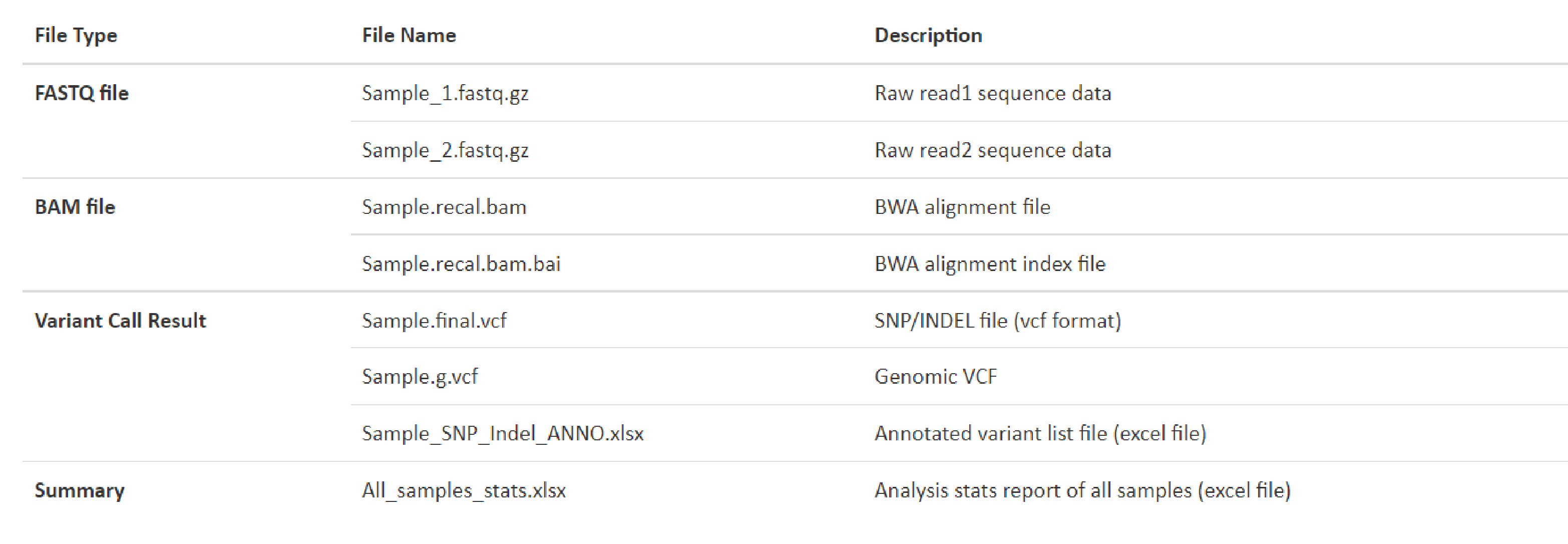
弊社html形式のレポートサイトの、DELIVERABLESの頁にRaw dataおよび解析結果のダウンロードリンクがあります。
Raw dataはデータベース登録時や、弊社でのデータ保管期間経過後に、追加データ解析をご希望の場合に必要なファイルとなります。
ダウンロードできる期間は約2週間となっておりますので、必ず全てのファイルをダウンロード頂きますよう、宜しくお願い致します。なお、2週間経過後も1ヵ月はデータを保管しておりますので、再度ダウンロードが必要な場合はngs@macrogen-japan.co.jpまでご連絡ください。
ダウンロードしたFastq.gzファイルもしくはHDD内のFastq.gzファイルは、ファイルの解凍前に”md5sum値”のご確認くだい。
”QuickHash-GUI”という、フリーのアプリケーションもございます。https://www.quickhash-gui.org/downloads/
もしお手持ちのソフトウェアが無ければこちらをご取得下さい。
QuickHash-GUIでのmd5sum値の確認方法は下記となります。
“QuickHash-GUI.exe”アプリケーションを起動します(例1)。
①“FileS”タブをクリックし、
②“Algorithm”、”MD5”を選択してください。md5sum値を確認したいfastqファイルが入っているフォルダを
③“Select Directory” から選択していただきますと、自動的に解析が進行致します。
出力は、csvファイルまたはtxtファイルとして保存することができます。
使用するシステムの性能により、処理に時間がかかる場合がございます。表示された数字と、レポートに記載のmd5sum値の一致を確認できましたら作業完了となります。あわせてQuickHash-GUIのユーザーマニュアルもご確認ください(例2)。
例1 QuickHash-GUI起動画面
例2 QuickHash-GUIfile中身
※本”納品レポートの見方”中のデータは一般的な納品例となっております。
お手元のレポートと一致しない場合もございますので、ご了承ください。
Experimental / Analysis Methods and Workflow
こちらの項目では、ライブラリ作製からシーケンス解析までを”Experimental” 、得られたリードデータを基に実施したデータ解析を”Analysis”と分類し、各々のワークフローを簡単に記載しています。
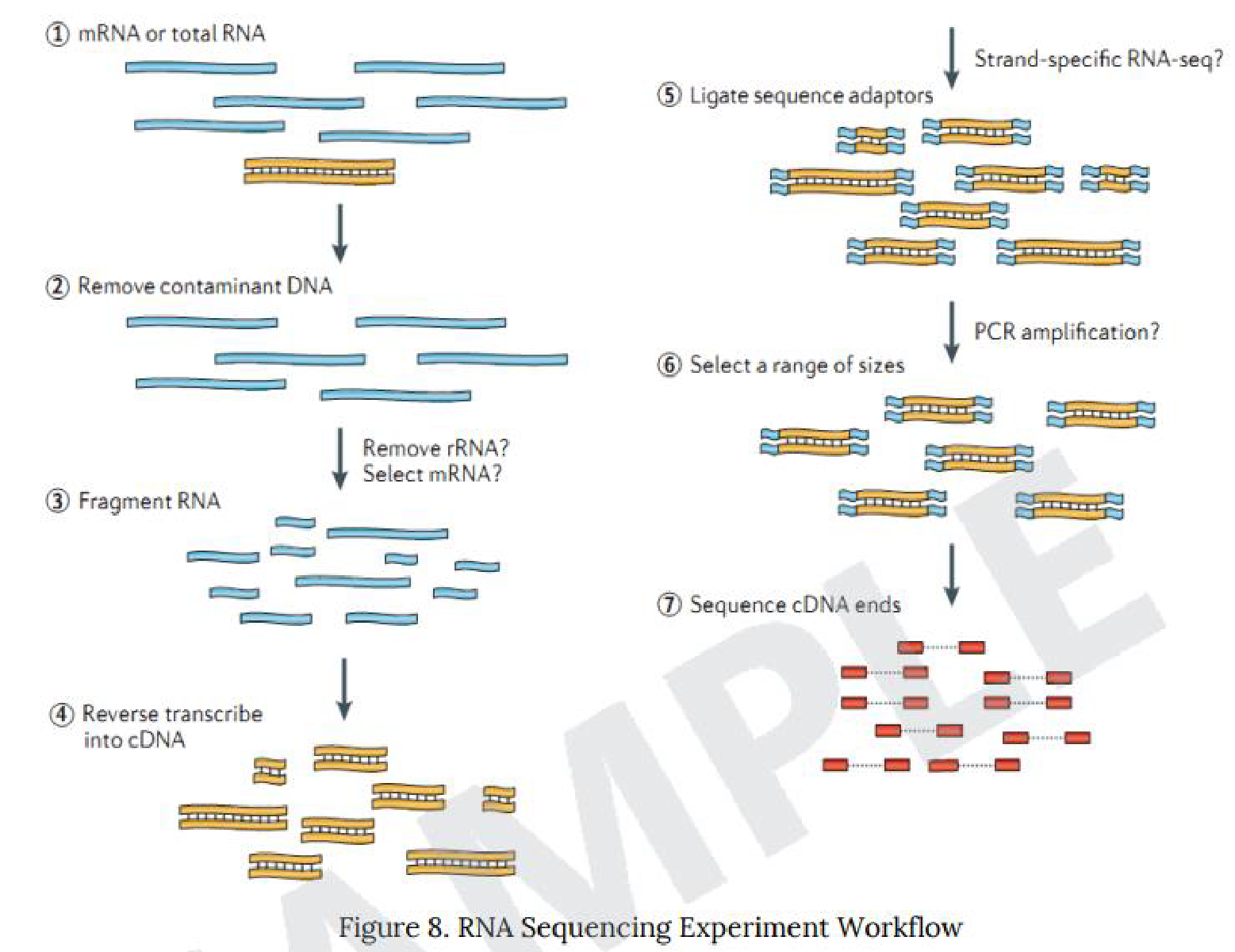
例1 RNA Sequencing Experiment Workflow

例2 Analysis Workflow
Summary of Data Production
こちらの項目では得られたリードデータのリード数、GC%、Qualityの確認結果を記載しています。

例1 Raw data stats
・Phredというプログラムで算出したQuality Score(QS)
= Phredクオリティスコアベースコールにおけるエラー率の予測指標
Q20: Phred QSが20以上の塩基の割合
Q30: Phred QSが30以上の塩基の割合
※QSの詳細は9. Appendixにも記載があります。
例2 Read quality at each cycle of sample
FASTQCで算出したQSを基に、Forward(read1)およびReverse(read2)について、リードの位置ごとのQSを図示しています。
・縦軸:QS、横軸:リード上での位置
緑色領域:Good Quality、黄色領域:Acceptable Quality、
赤色領域:Bad Quality を示しており、得られたリードを平均して評価した際に、どのQualityにあたるのか確認できます。
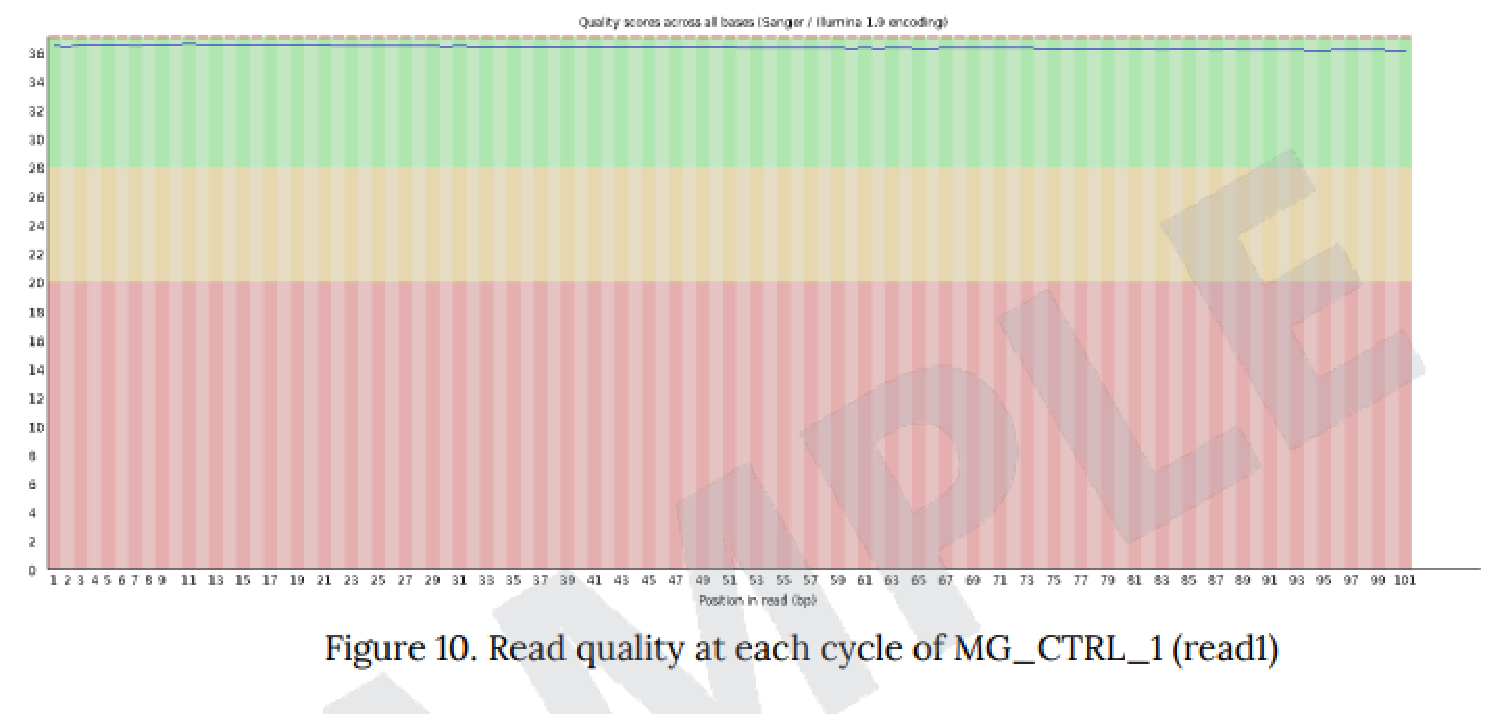

例2 Read quality at each cycle of sample
FASTQCで算出したQSを基に、Forward(read1)およびReverse(read2)について、リードの位置ごとのQSを図示しています。
・縦軸:QS、横軸:リード上での位置
緑色領域:Good Quality、黄色領域:Acceptable Quality、
赤色領域:Bad Quality を示しており、得られたリードを平均して評価した際に、どのQualityにあたるのか確認できます。
Reference Mapping and Assembly Results
こちらの項目では得られたリードデータをリファレンス情報にマッピングし、マッピングの状況から算出した、既知転写産物および遺伝子の発現量のリストおよび新規転写産物として予測した転写産物の発現量算出結果リストを確認できます。

例1 Known transcripts Expression Level
リードカウント、Transcript Length および発現量など、得られた情報をまとめた一覧をExcel 形式でまとめています。こちらのリストは
Result_RNASeq_Excel フォルダ内の
Expression_profileに以下名称にて保管されています。
・Expression_Profile.GRCh38.transcript.xlsx
・Expression_Profile.GRCh38.genes.xlsx
例2 新規転写産物の分類
図示されておりますSplicingパターンの取り方を基に、
計13パターンに分けて評価分類しています。
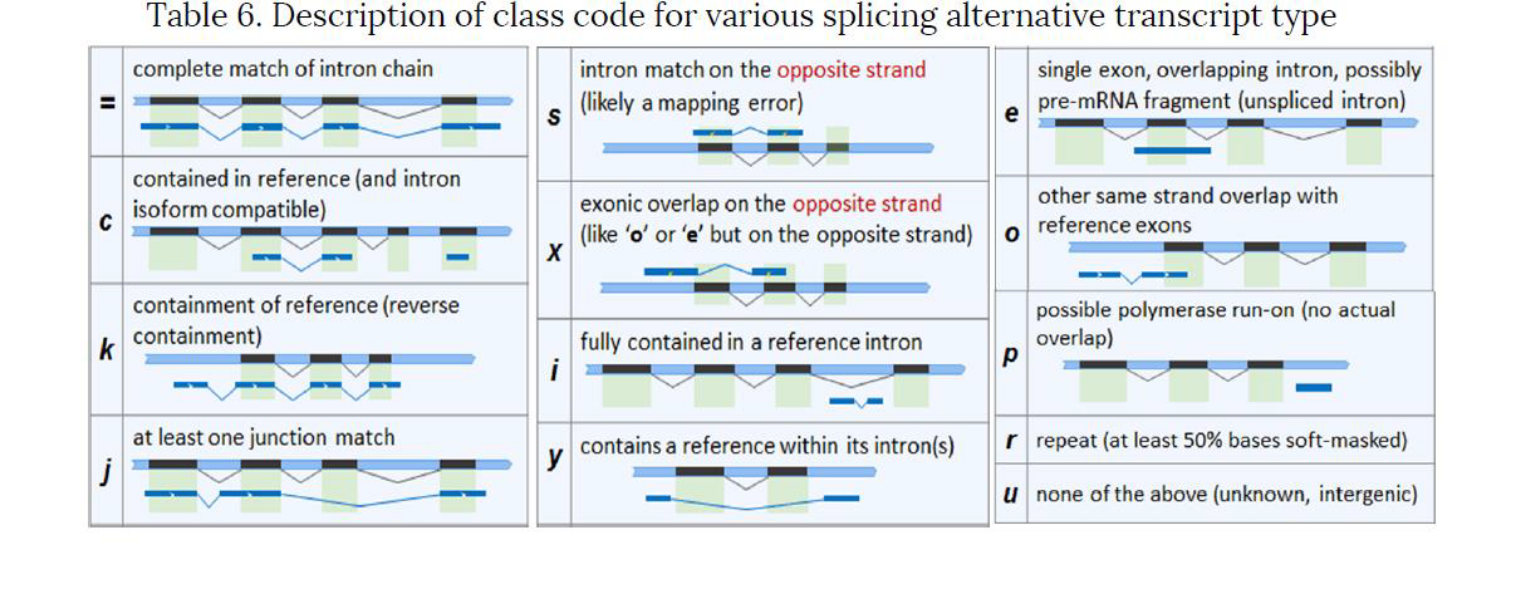
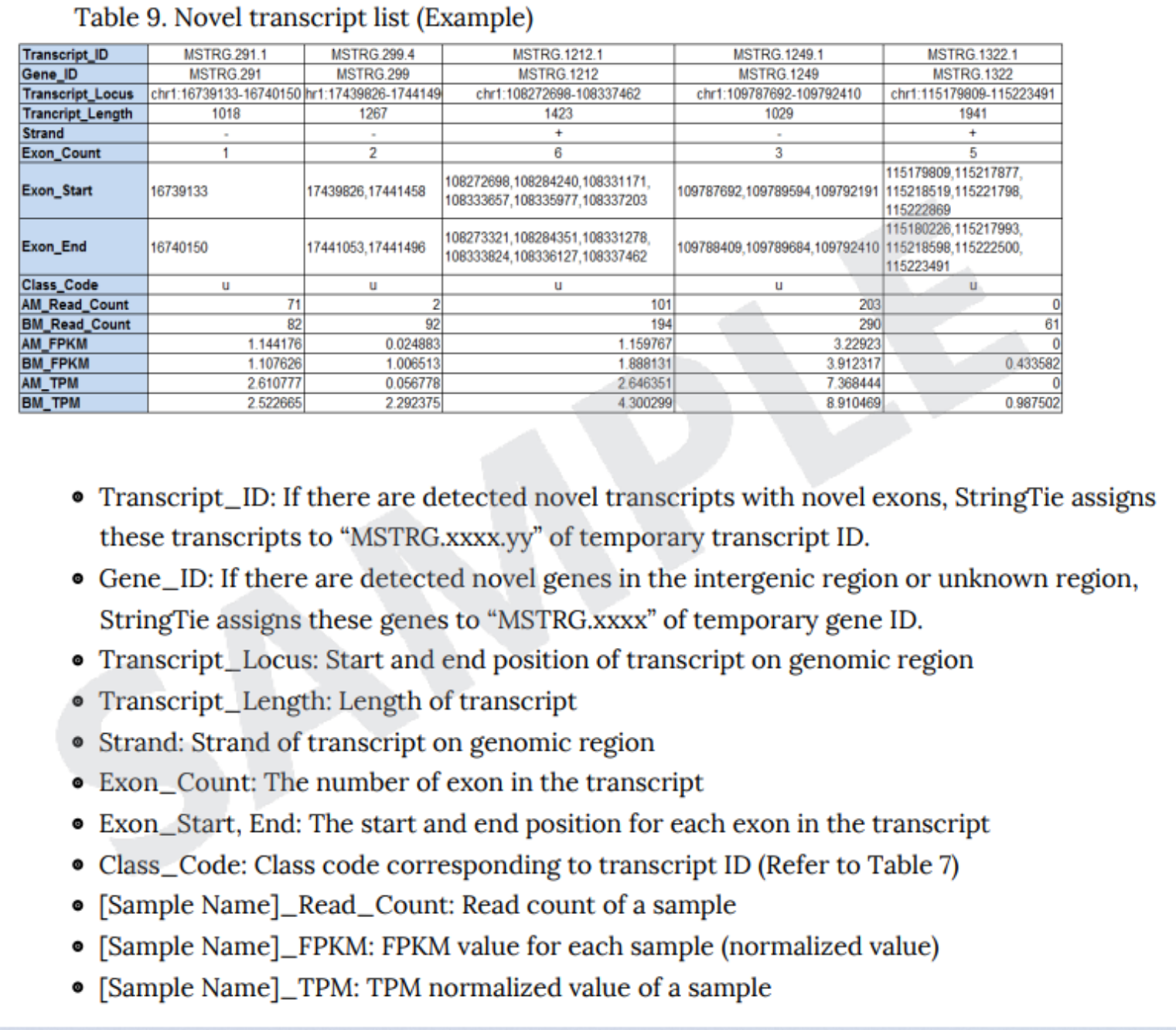
例3 Novel Transcript list
例2での分類結果、リードカウント、Exon数および発現量など、本解析で得られた情報をExcel形式にまとめています。
ファイルはResult_RNASeq_Excelフォルダ内の
Novel Transcript Analysisに以下名称にて保管されています。
- Expression_Profile_with_Novel.GRCh38.transcript.xlsx
※こちらは転写産物のリストとなります。
- Expression_Profile_with_Novel.GRCh38.genes.xlsx
※こちらは遺伝子でのリストとなります。
Differentially Expressed Gene Analysis Results
こちらの項目の解析結果の内容は
「Result_RNASeq_Excel 」「 DEG_Result 」に格納されている、
レポートファイル“Analysis_Result.html”にまとめています。
こちらのレポートファイルと併せてご確認ください(例 1) 。
以下6 項目に関してこれ以降でご案内します。
①遺伝子のフィルタリング
②各サンプルの相関関係の把握
③発現変動遺伝子の分布状況の確認
④Heatmap Analysis
⑤GO Enrichiment Analysis
⑥KEGG Enrichiment Analysis
※解析希望時のみ、有償オプション。
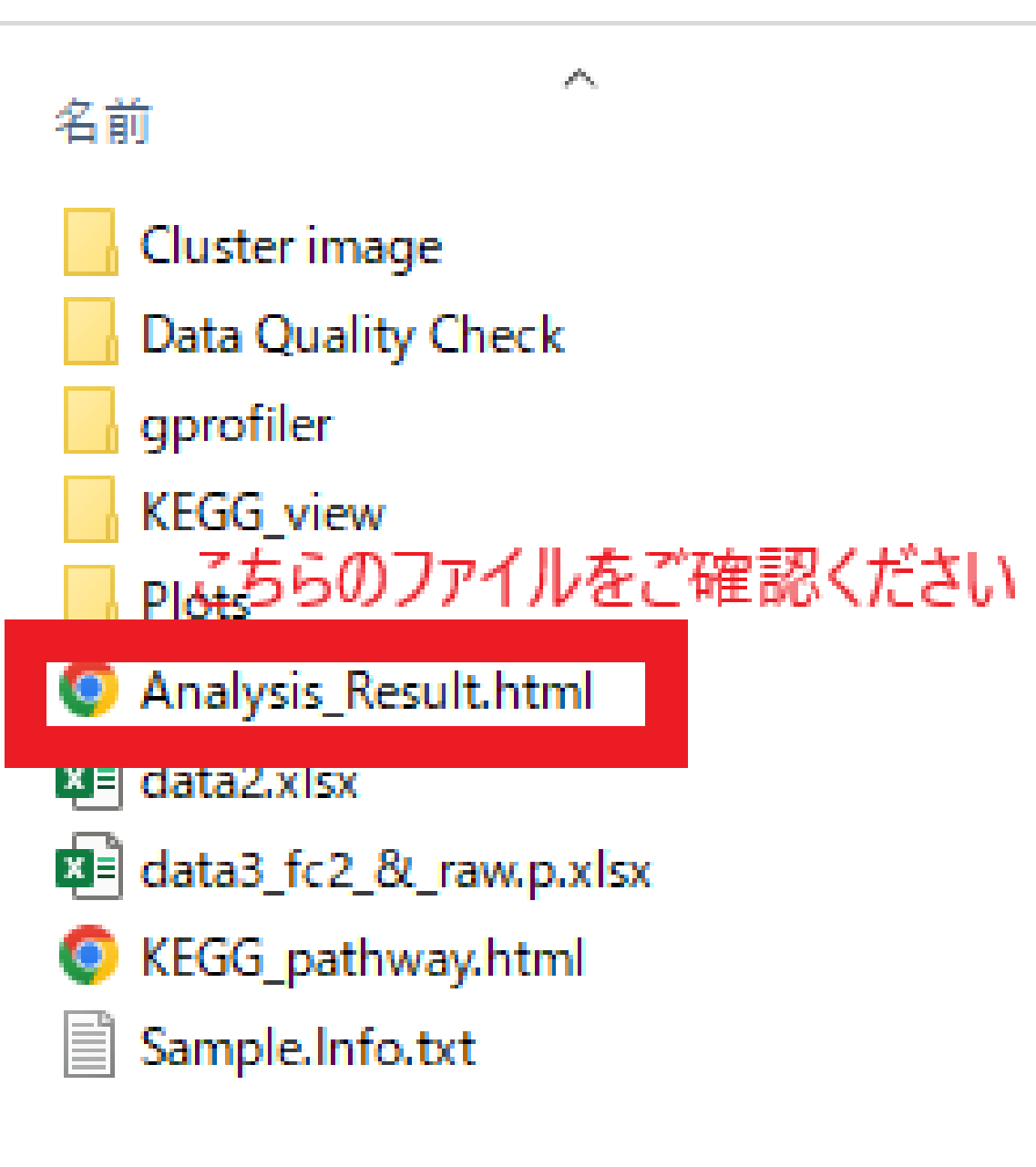
例1 Analysis_Result.html について
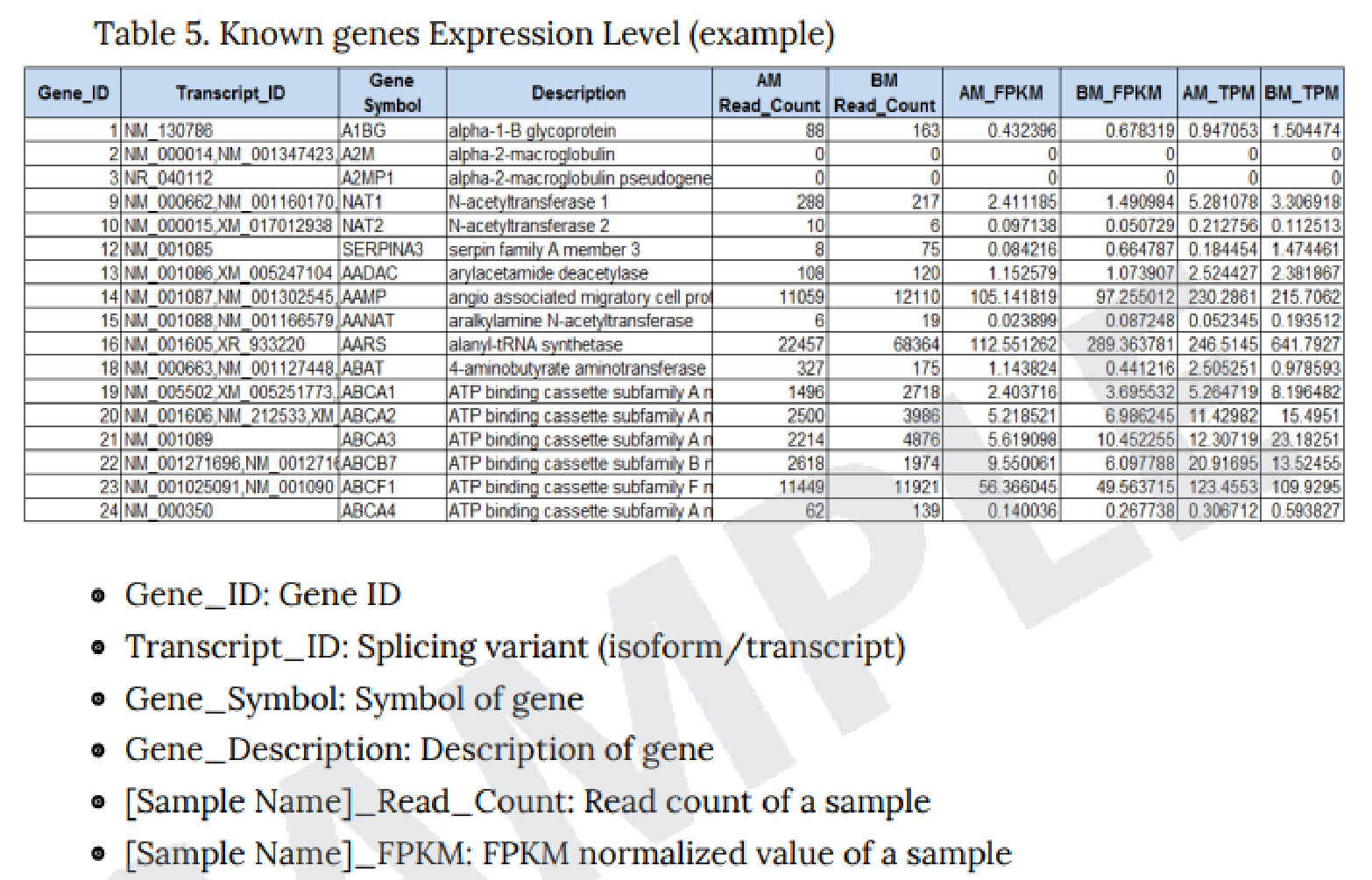
例2 Expression Profile.GRCh38.gene.xlsx
データ解析で使用しております数値データのRaw data は以下の名称でフォルダに格納されています。
・Raw data : Expression Profile.GRCh38.gene.xlsx
※ “result_RNAseq_excel” 内の ”Expression Profile”フォルダに格納。
・Normalization 後の Raw data : data2.xlsx
・FC 値、 P 値を加味した統計データ : data3_fc2_&_raw.p.xlsx
全比較内容のうち少なくとも1 つにおいて、|fc|≧ 2 、raw.p < 0.05 の条件を満たす遺伝子をフィルタリングした結果をまとめたものが、”data3_fc2_&_raw.p.xlsx “となります。
例3 data3_fc2_&_raw.p.xlsx

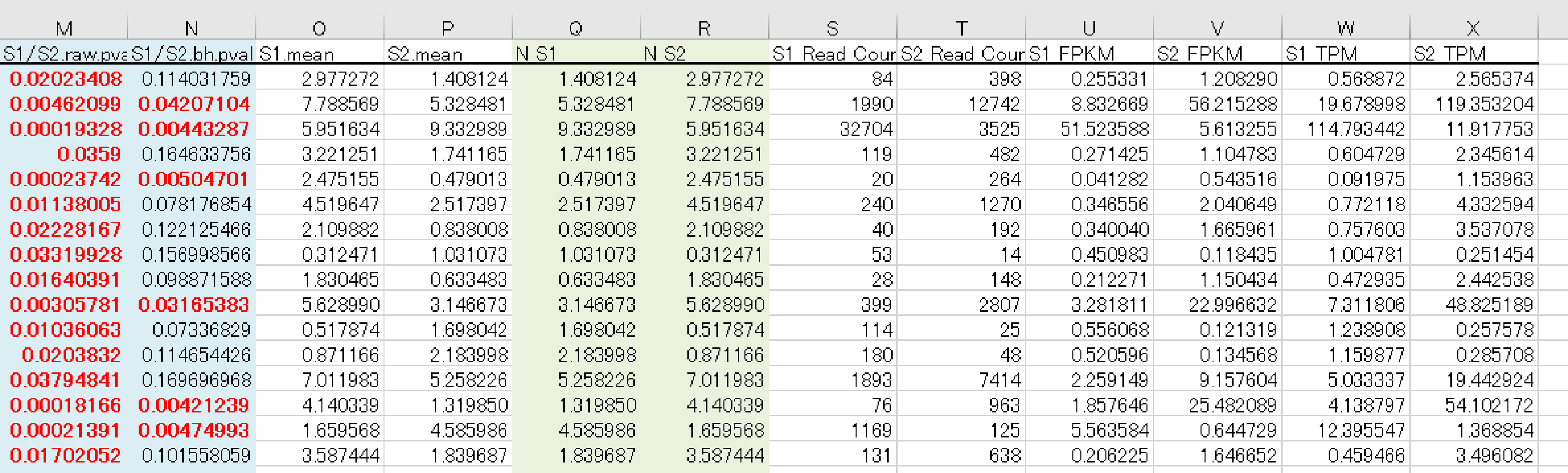
全比較内容のうち少なくとも1 つにおいて、|fc|≧ 2 、raw.p < 0.05 の条件を満たす遺伝子をフィルタリングした結果をまとめたものが、”data3_fc2_&_raw.p.xlsx “となります。
【カラム項目詳細 】
- A 列: Gene ID
- B 列: Transcript ID
- C 列: Gene Symbol
- D 列: データベース記載の遺伝子説明
- E 列: 遺伝子の分類
- F 列: Protein ID
- G 列: HGNC(HUGO Gene Nomenclature Committee) ID
- H 列: MIM(Mendelian Inheritance in Man) ID
- I 列: Ensembl ID
- J 列: IMGT/GENE DB ID
- K 列: Test/Control での Fold Change(FC) 値
- L 列: Test/Control での log Counts per Million reads (CPM) 値
- M 列: Test/Control での P 値
- N 列: Test/Control での FDR correction P 値
OーX 列につきましては、各サンプルごとの平均値、 raw signa の TMM 補正値、リードカウント、FPKM 値、 TPM 値を記載しています。
※詳細は“ Analysis_Result.html”“ III-5. Column Information ”にございます。
こちらのExcel ファイルに GO Enrichiment Analysis 、 KEGG Enrichiment Analysis ※オプション の結果も別シートで記載していますので、あわせてご確認ください。
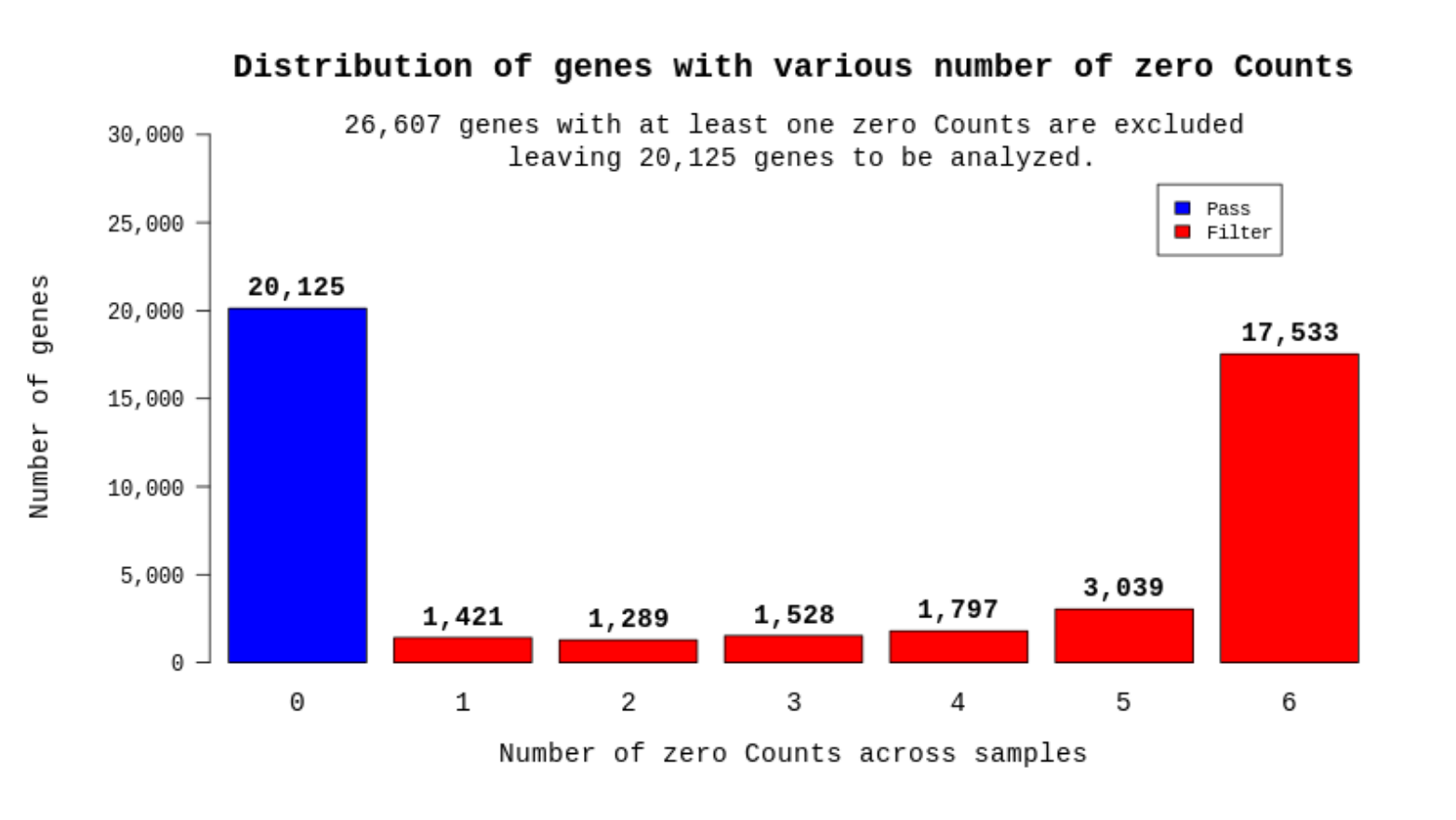
例1 Distribution of genes with various number of zero counts
いずれかのサンプルでリードカウントが0となった遺伝子を解析対象から外すゼロカウントフィルタリングを行います。図ではサンプルデータごとにリードカウント=0の遺伝子をフィルタリングし、サンプル数を跨いで0となる遺伝子数を表示しています。全てのサンプルで発現している遺伝子のみ(青Box)、以降の解析では使用しています。
※Raw dataのExcelにはリードカウントが0の遺伝子も記載しています。
ご希望いただいた場合、0カウントフィルタリングの条件を変更して、再解析も可能ですので、ご希望の場合は担当者にご連絡ください。
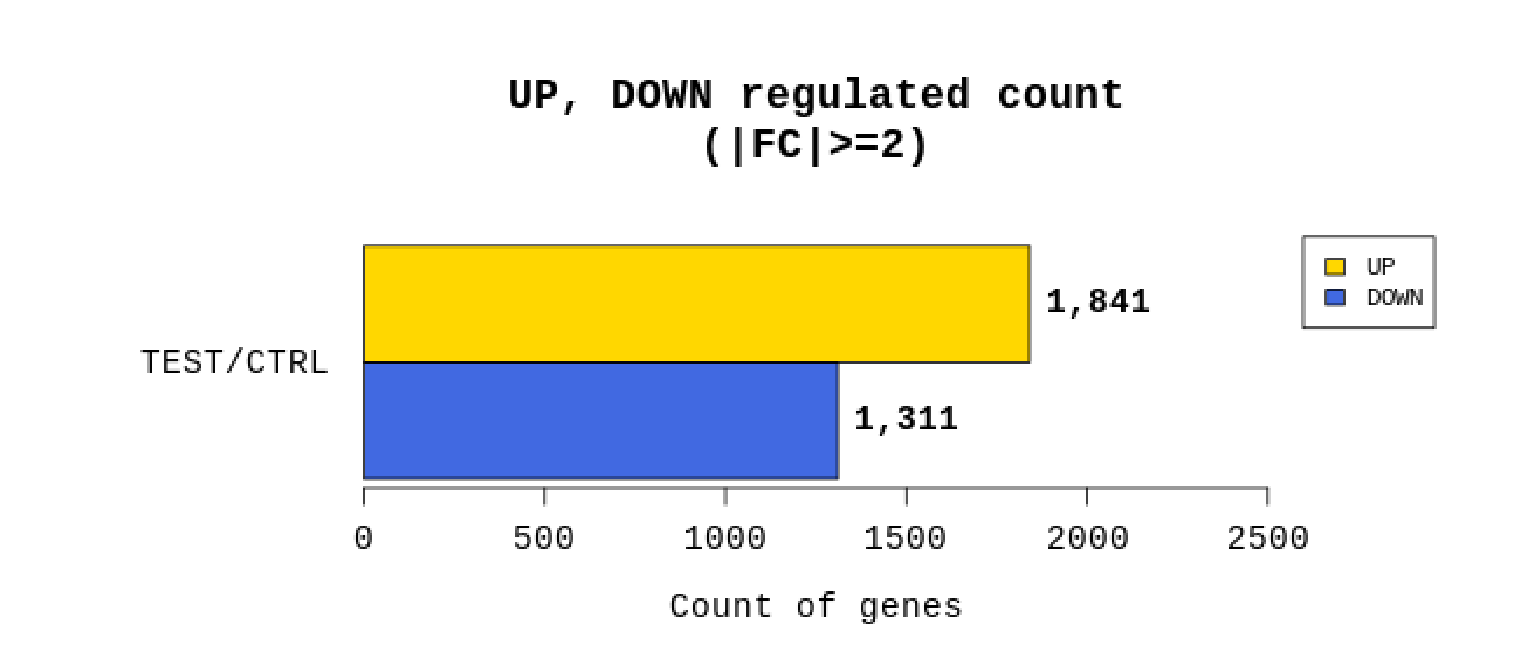
例2 Whole genome resequencing analysis process
リードカウントを基にTest/Controlで計算しFold change(FC)を算出。あわせてp値も算出し、|FC|≧2かつp値 < 0.05の遺伝子をフィルタリングしています。
(フィルタリングされる遺伝子数が極端に少ない場合は|FC|≧1.5にて、フィルタリングを行う場合もあります。)
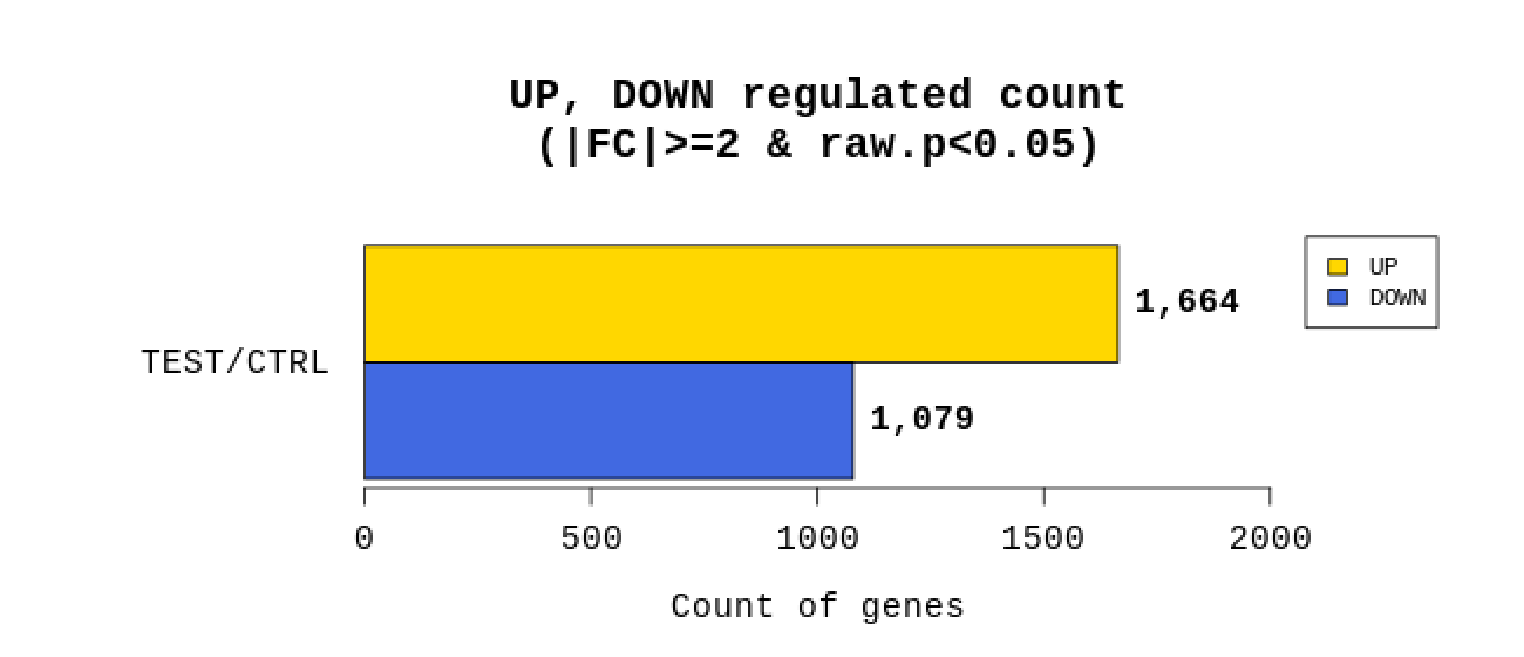
例2 Whole genome resequencing analysis process
リードカウントを基にTest/Controlで計算しFold change(FC)を算出。あわせてp値も算出し、|FC|≧2かつp値 < 0.05の遺伝子をフィルタリングしています。
(フィルタリングされる遺伝子数が極端に少ない場合は|FC|≧1.5にて、フィルタリングを行う場合もあります。)
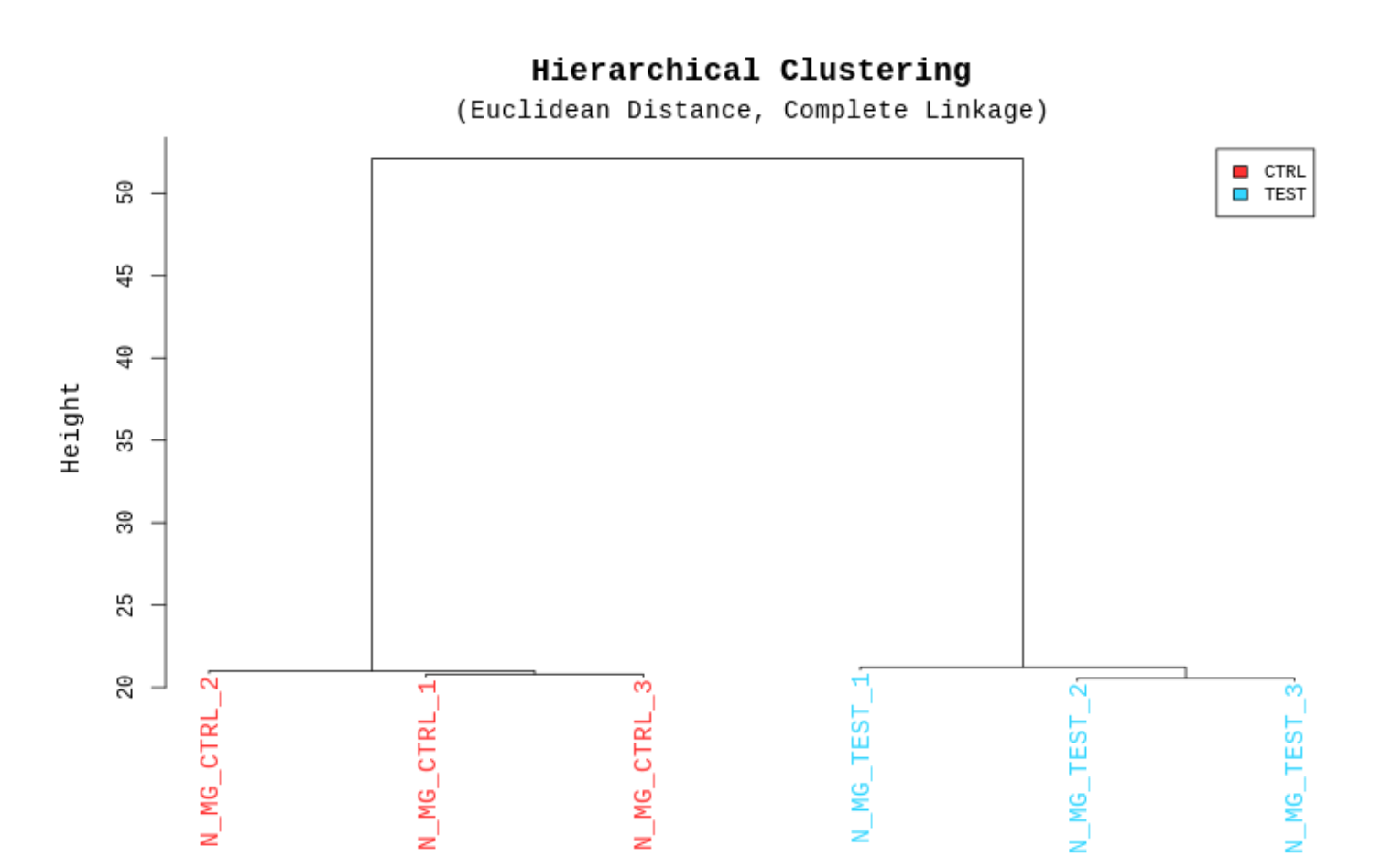
各サンプルの相関関係を複数の図表で表しています。
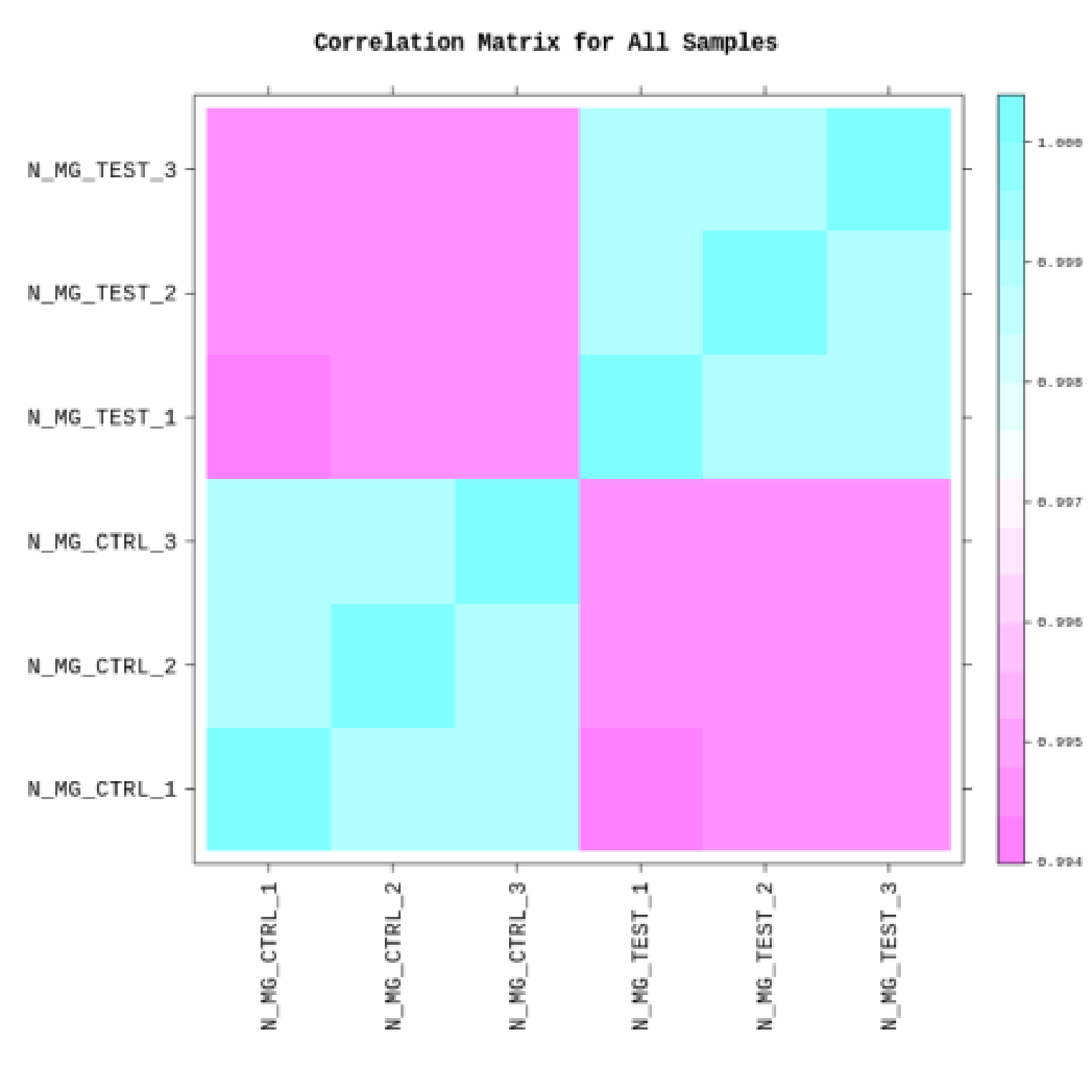
例1Correlation Matrix for All Samples
サンプルデータごとにピアソンの積率相関係数をとり、ヒートマップ化しています。
例2 Hierarchical Clustering Analysis
サンプルデータごとのノーマライズ値を元に、発現の類似性を階層的に表しています。

例3 Multidimensional Scaling Analysis
二次元プロットにより、発現量のばらつきとサンプルとの相関を表しています。
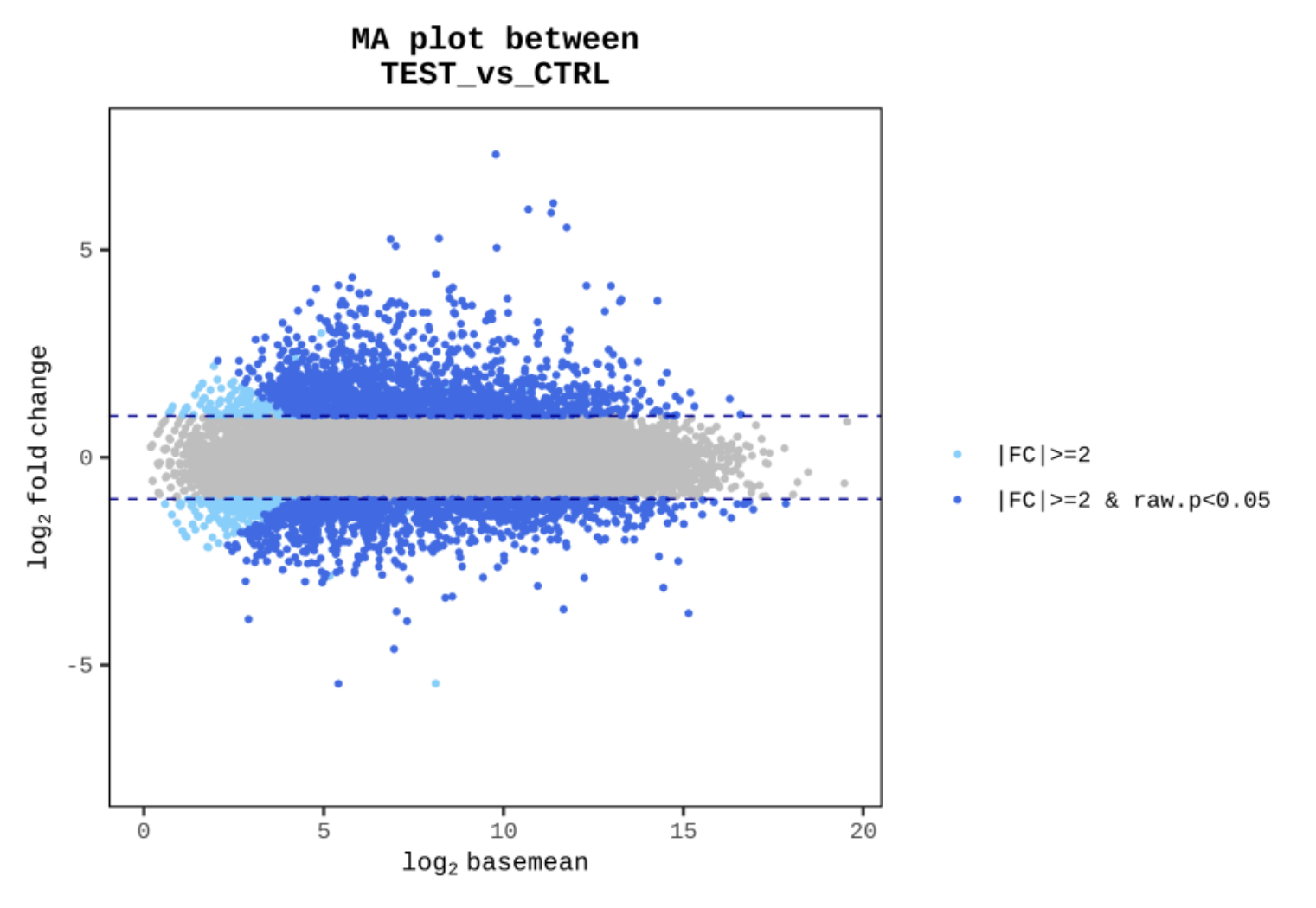
|FC|≧2かつp値 < 0.05の遺伝子に対して各種Plotを作図しています。
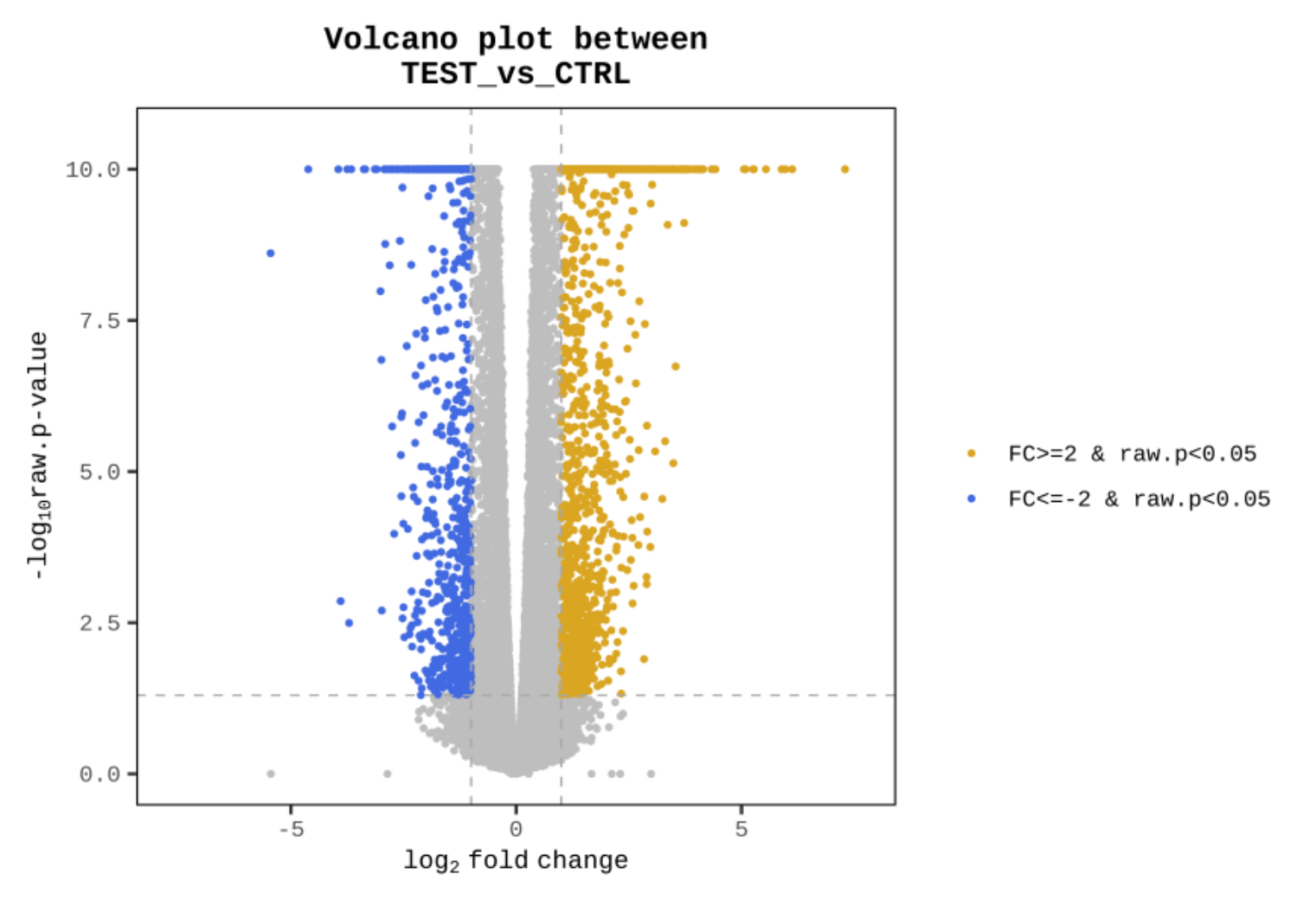
例1 Volcano Plot between TEST vs CTRL
縦軸をp値の対数、横軸をFC値の対数として分布図化することで、|FC|≧2かつp値 < 0.05の遺伝子の(青色(-), 黄色(+))の分布を表しています。
※n≧2の際に作成されます。
例2 MA Polt between TEST vs CTRL
縦軸をFC値の対数、横軸を発現量の相乗平均
として分布図化することで、
|FC|≧2かつp値 < 0.05の遺伝子(水色、青色)の分布を表しています。
※解析結果によっては|FC|≧2が条件でない場合もございます。
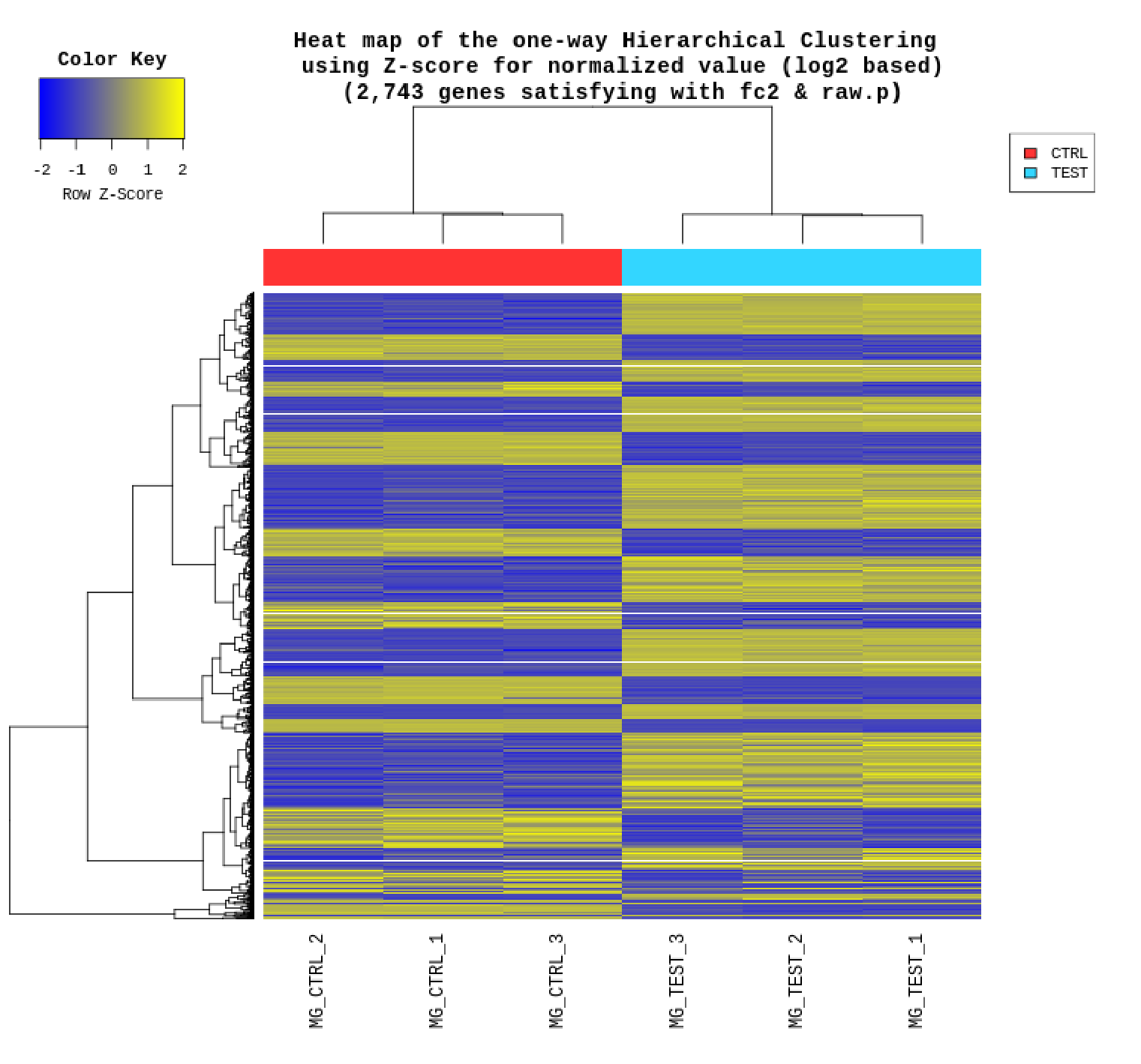
例1 Heatmap
|FC|≧2かつp値 < 0.05の遺伝子に対してあわせてノーマライズしたすべてのサンプル及び各比較パターンごとにHeatmapを作成しています。
Gene Ontology(http://geneontology.org/) database登録情報より、発現量変動が大きかった遺伝子がBiological Prosess (BP), Cellular Component (CC), Moleculer Function (MF)の3つの観点からどのような機能グループ(GO term)に属するかを分類し、発現量変動の大きかったGO termの一覧表及びPlotを作成しています。
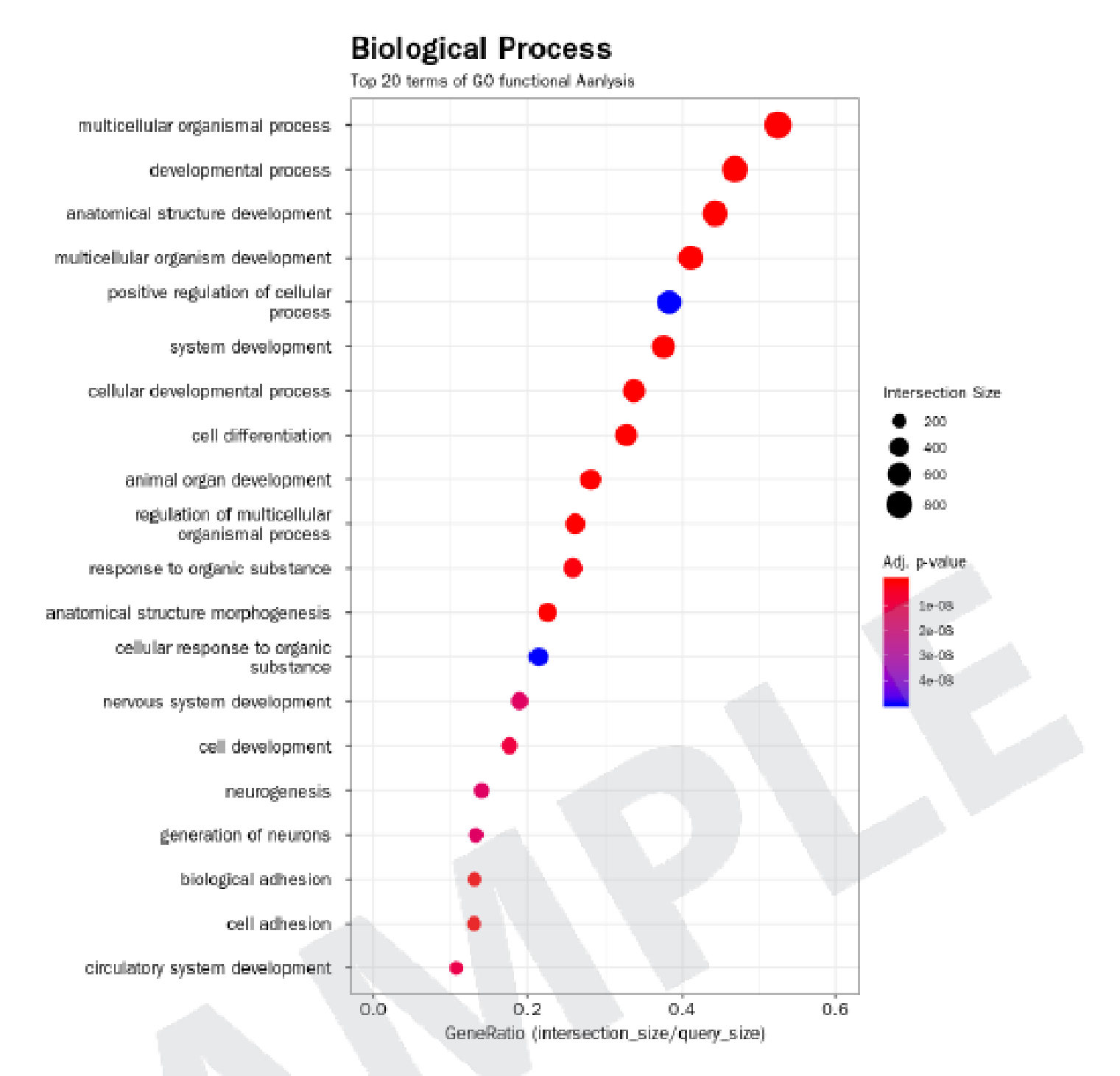
例1 Biological ProcessにおけるGO解析結果
検定によるp値< 0.05, 0.01, 0.001における、上位20位までの遺伝子をグラフ化しています。
- 縦軸:遺伝子系統名
- 横軸:全体の遺伝子数における該当遺伝子数の割合
- ●:遺伝子数の割合
- カラーバー:p値
「result_RNAseq_excel」→「DEG_result」→「gprofiler」内に各種Plotが格納されてます。比較パターンごとのフォルダ内には“gprofiler.bar.png”および“gprofiler.png”がありますが、同一内容の図表の表示の仕方を棒グラフ(.bar.png)とドットプロット(.png)で表しています。例1はドットプロットでの図表となります。
【sizefilt.png につきまして 】
格納されております“gprofiler.sizefilt.png” は、p-value が 0.05 未満かつ term_size が 10~500 のもののみから作成された Plot になります。
GO termは term size( その term に含まれる遺伝子数)が様々です。非常に大きいまたは小さいGO term は、計算方法によっては、統計的有意性が大きく算出されることがあります。そのため、通常の解析結果と合わせて、term size が 10-500 にフィルタリングして、別途作成した Plot も納品しています。
例2 GO_statSheet
データ解析で使用しております数値データのRaw data は
以下の内容で“data3_fc2_&_raw.p.xlsx” に記載しています 。
・GO_stats
・GO_genes
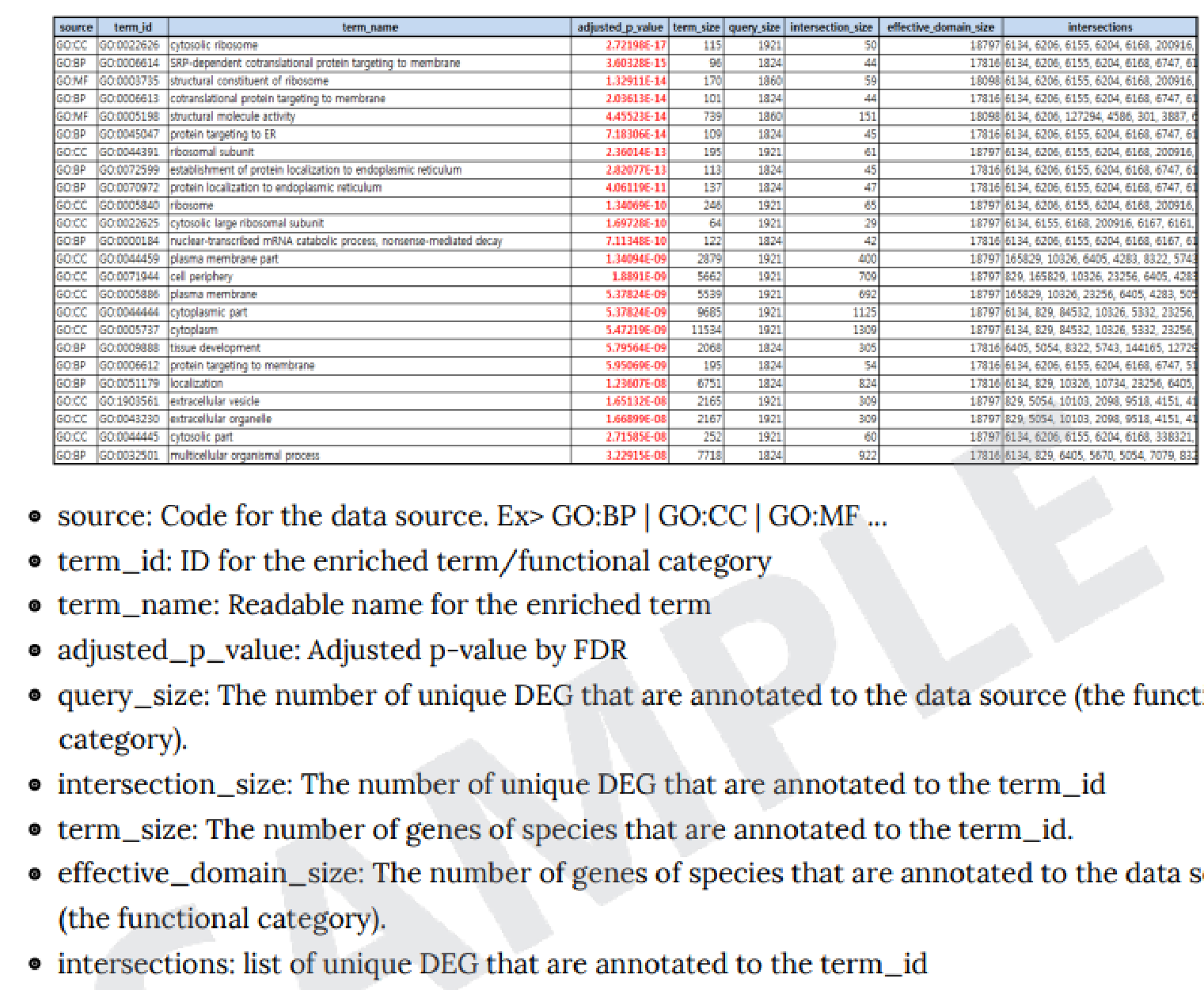
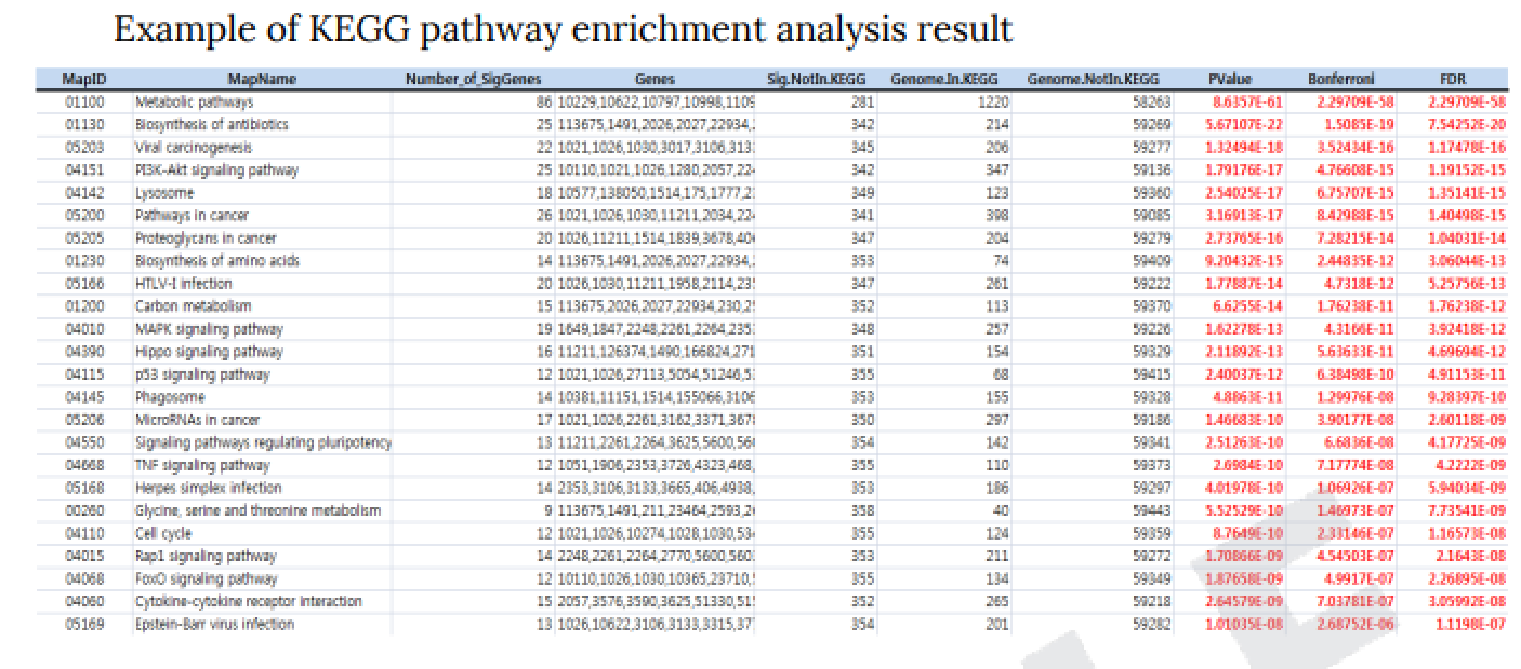
KEGG(https://www.genome.jp/kegg/kegg_ja.html)というdatabaseの情報に、各比較パターンでの発現量変動を当てはめることで、各Pathway 上でどのような遺伝子に有意な変化があったかを図示しています。
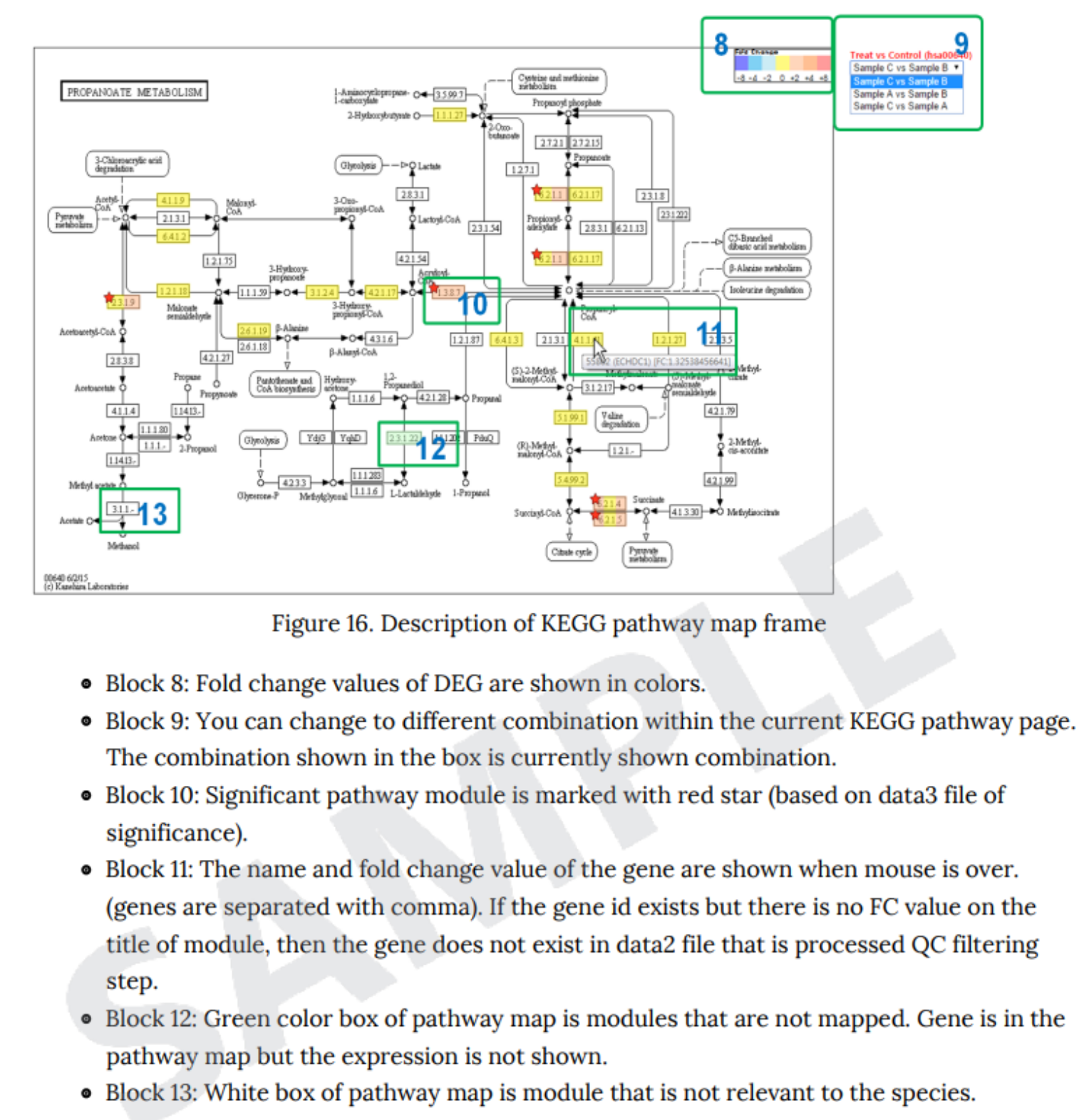
例1 Description of KEGG pathway map frame
各Pathwayで有意差のあった遺伝子の発現量比を8.のような色分けでPathwayマップ図内に表しています。
また、各pathwayで重要なモジュールには★マークを付けています。
※Pathwayマップは KEGG データベースに登録の情報を使用しているため、お客様の希望に合わせての作図・改定の対応は致しかねます。
例2 KEGG_stat Sheet
データ解析で使用しております数値データのRaw data は
以下の内容で“data3_fc2_&_raw.p.xlsx” に記載しています 。
・GO_stats
・GO_genes
SNP and Indel Discovery
※有償オプション。生物種によっては対応できないため事前にご相談ください。
※有償オプション。生物種によっては対応できないため事前にご相談ください。
シーケンス結果をリファレンス情報にマッピング、SNV calling 後、Annotationを付与した結果を納品します。
解析結果は“SNV_Call サンプル名.xlsx”の Excel 形式にて格納されています。

例1 SNV 納品結果例
各項目の詳細につきまして、PDF ファイルにまとめがあります。
以下で示しておりますリンクが納品物 PDF にありますので、こちらよりご取得・ご確認ください。

Fusion Gene Prediction Results
※有償オプション。生物種によっては対応できないため事前にご相談ください。
※有償オプション。生物種によっては対応できないため事前にご相談ください。
以下3種類のProgramを使用した予測結果を納品しています。
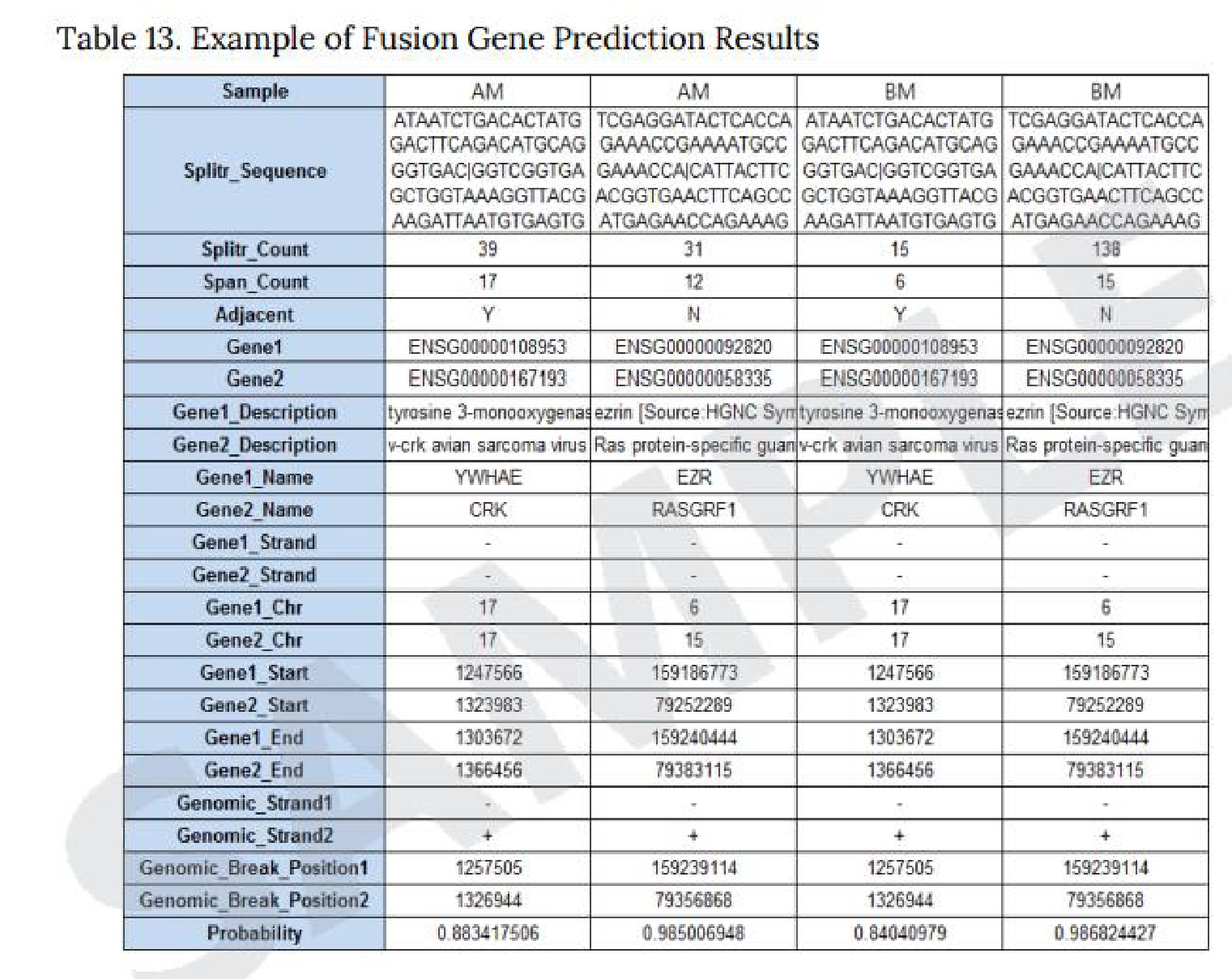
● Defuse program
Defuseprogramを使用し、マッピング状況から2遺伝子間を跨ぐリードを抽出し、融合遺伝子を予測します。
※左上の図はExcel形式での納品例。
● Fusion Catcher
Fusion Catcherを使用し、Mapping状況から既知融合遺伝子であるか否か、また、転座、キメラかを予測します。
※右上の図はExcel形式での納品例。
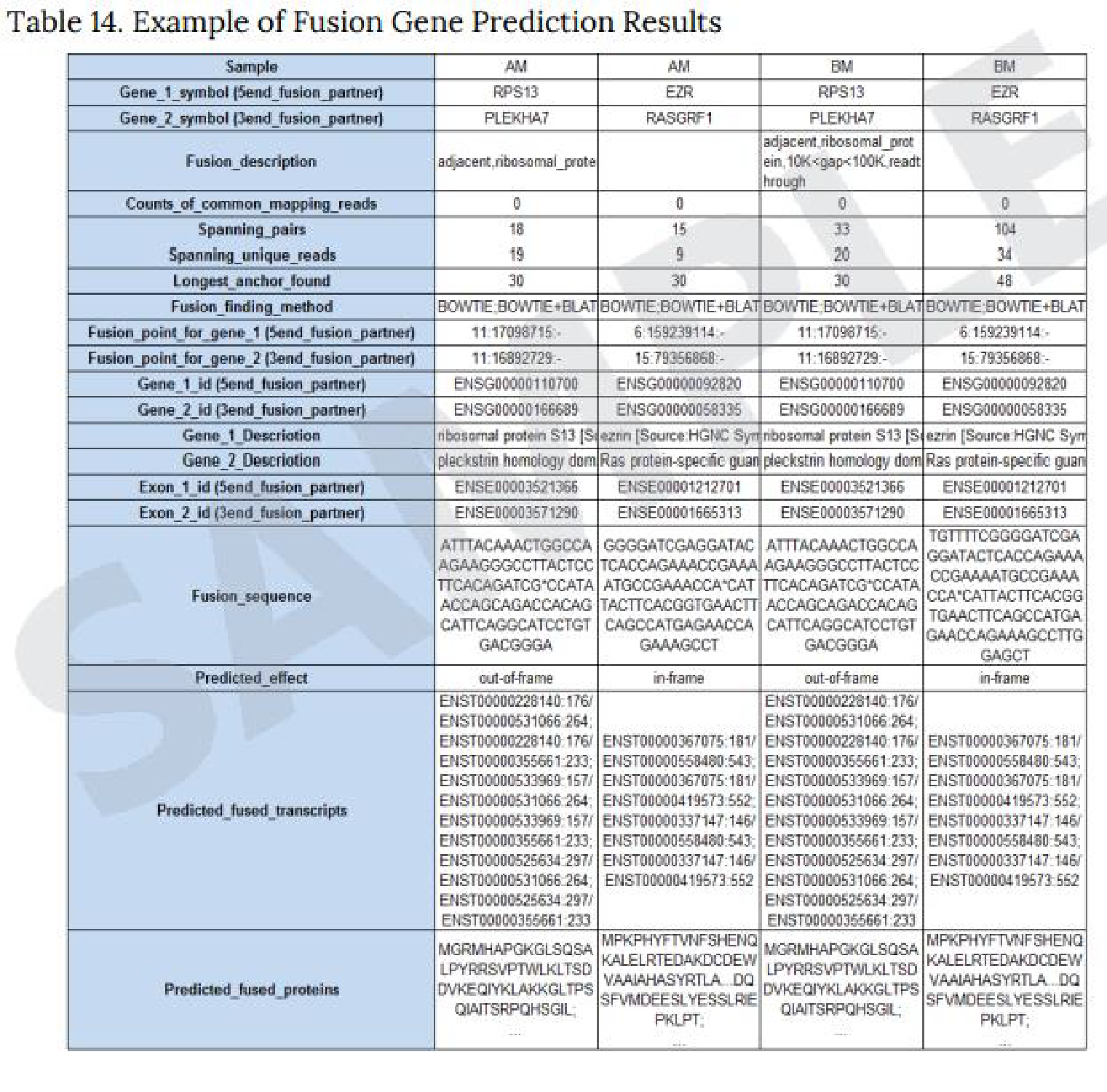
● Arriba
Arribaを使用し、 STAR aligner でのマッピング状況から融合遺伝子を予測、および融合までの動態を図示しています。
※Arribaが使用できるのは human のみとなります。
※PDFファイル、 Excel 形式での納品。
-
融合遺伝子形成までの模式図
※染色体情報、 Coverage, 、転写様式、配列、BreakPointの内容が確認できます。 -
Circos Plot
※各染色体ごとに 全 ゲノム情報を環状に表記し、融合している部位を線つなぎで表示しています。 -
予測融合遺伝子のドメイン構造の模式図
※Excel形式での納品例(例4) 。
例3 Arriba による解析結果

例4
Data Download Information
Raw dataおよび解析結果のダウンロードリンクが記載されています。
Raw data(fastqファイル)は論文投稿時に行うデータベース登録時に、また、弊社でのデータ保管期間経過後に、追加データ解析をご希望の場合に必要なファイルです。
ダウンロードできる期間は約2週間となっておりますので、必ず期間中に全てのファイルをダウンロードして下さい。また、ダウンロードしたfastq.gzファイルはファイルに破損がないかの確認のため、必ず”md5sum値”の照合を行ってください。ご確認をお願いしております。
確認、照合方法につきましては”CAUTION”をご確認ください。
弊社pdf 形式のレポートファイルの、 Data Download Information の頁にRaw dataおよび解析結果のダウンロードリンクがあります。
Raw dataはデータベース登録時や、弊社でのデータ保管期間経過後に、追加データ解析をご希望の場合に必要なファイルとなります。
ダウンロードできる期間は約2週間となっておりますので、必ず全てのファイルをダウンロード頂きますよう、宜しくお願い致します。なお、2週間経過後も1ヵ月はデータを保管しておりますので、再度ダウンロードが必要な場合はngs@macrogen-japan.co.jpまでご連絡ください。
ダウンロードしたFastq.gzファイルもしくはHDD内のFastq.gzファイルは、ファイルの解凍前に”md5sum値”のご確認くだい。
”QuickHash-GUI”という、フリーのアプリケーションもございます。https://www.quickhash-gui.org/downloads/
もしお手持ちのソフトウェアが無ければこちらをご取得下さい。
QuickHash-GUIでのmd5sum値の確認方法は下記となります。
“QuickHash-GUI.exe”アプリケーションを起動します(例1)。
①“FileS”タブをクリックし、
②“Algorithm”、”MD5”を選択してください。md5sum値を確認したいfastqファイルが入っているフォルダを
③“Select Directory” から選択していただきますと、自動的に解析が進行致します。
出力は、csvファイルまたはtxtファイルとして保存することができます。
使用するシステムの性能により、処理に時間がかかる場合がございます。表示された数字と、レポートに記載のmd5sum値の一致を確認できましたら作業完了となります。あわせてQuickHash-GUIのユーザーマニュアルもご確認ください(例2)。
例1 QuickHash-GUI起動画面
例2 QuickHash-GUIfile中身
※本”納品レポートの見方”中のデータは一般的な納品例となっております。
お手元のレポートと一致しない場合もございますので、ご了承ください。
Raw_dataフォルダ内の report.html が Raw Data に関する報告書です。
Order情報や得られた Raw Data に関する情報、 fastq ファイルのダウンロードリンクが含まれます。

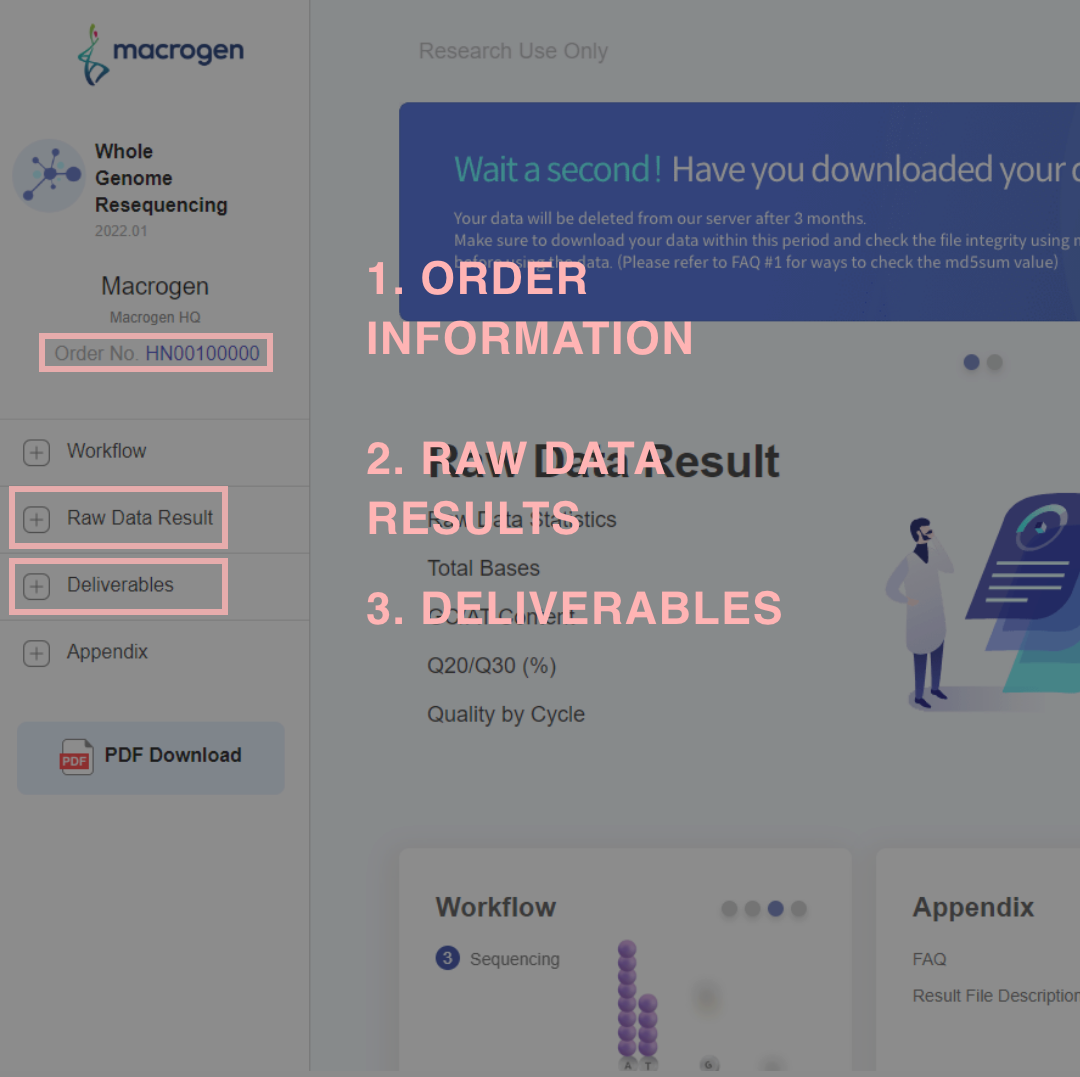
ORDER INFORMATION
本案件で使用したライブラリ調製キット、シーケンサーを記載しています。シーケンサー機種名について、
下記例では“illumina system”と記載がありますが通常弊社でASV 解析をご依頼の場合 MiSeq シーケンサーを使用しており、レポートではType of ReadはPaired-end 、 Read Length は301と表記されます。
illumiina MiSeq System
https://jp.illumina.com/systems/sequencing platforms/miseq.html
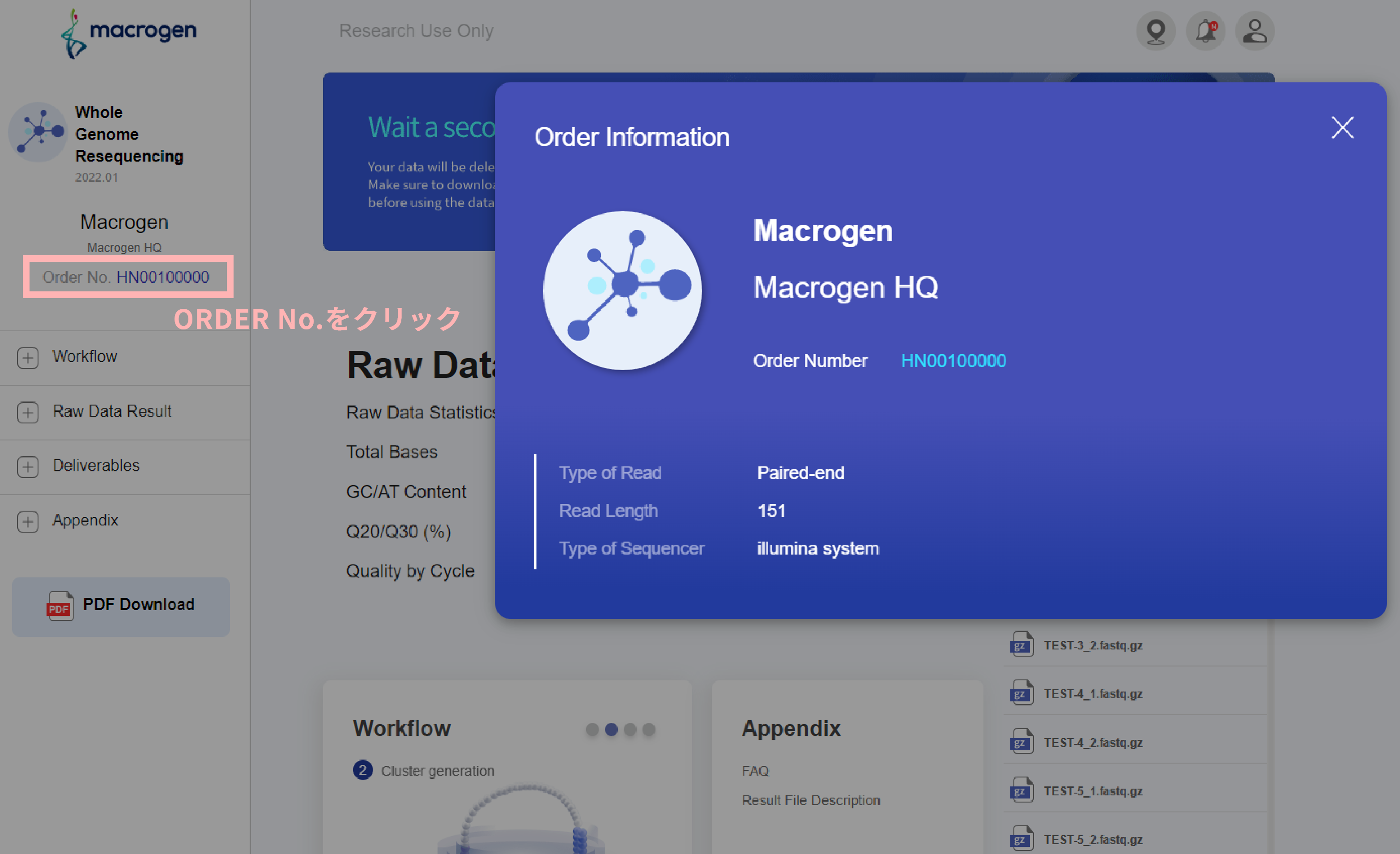
【Order Informationの例】
レポート左部のOrder No をクリックすると、Order Informationが表示されます。

RAW DATA RESULTS
得られたリードのリード数、GC %、 Quality を記載しています。

例1 Raw Data Statistics
Phredというプログラムで算出した Quality Score(QS)
=Phredクオリティスコアベースコールにおけるエラー率の予測指標
Q20(%):Phred QSが20以上の塩基の割合
Q30(%):Phred QSが30以上の塩基の割合
※QSの詳細は Appendix の Result File Description にも記載があります。
例2 Quality by Cycle
FASTQCというプログラムで算出した QS を基に、Forward(read1)および Reverse(read2) について、リードの位置ごとの QS を図示しています。
緑色領域:Good Quality 、黄色領域: Acceptable Quality 、赤色領域: Bad Quality を示しており、
得られたリードを平均して評価した際に、どのQuality にあたるのか確認できます。
縦軸:QS 横軸:リード上での位置
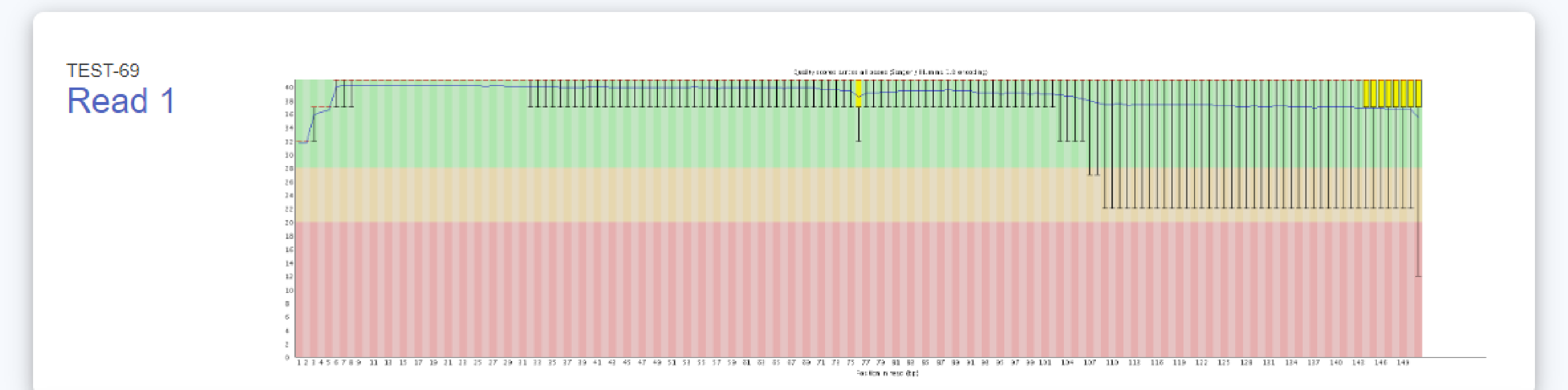
例2 Quality by Cycle
FASTQCというプログラムで算出した QS を基に、Forward(read1)および Reverse(read2) について、リードの位置ごとの QS を図示しています。
緑色領域:Good Quality 、黄色領域: Acceptable Quality 、赤色領域: Bad Quality を示しており、
得られたリードを平均して評価した際に、どのQuality にあたるのか確認できます。
縦軸:QS 横軸:リード上での位置
DELIVERABLES
Raw dataは論文投稿時に行うデータベース登録時に、また、弊社でのデータ保管期間経過後に、追加データ解析をご希望の場合に必要なファイルです。
ダウンロードできる期間は約2週間となっておりますので、必ず期間中に全てのファイルをダウンロードして下さい。
また、ダウンロードしたfastq.gz ファイルはファイルに破損がないかの確認のため、必ず”md5sum 値”の照合を行ってください。
確認、照合方法につきましては末尾”CAUTION” をご確認ください。
【fastqファイルダウンロードページの例】
ファイル名をクリックすると、ダウンロードが開始されます。

「受託番号_ASV 」フォルダ内のReport.html が ASV 解析に関する報告書です。 解析結果の生データや図表、解析のフローをまとめています。※下記納品フォルダの例
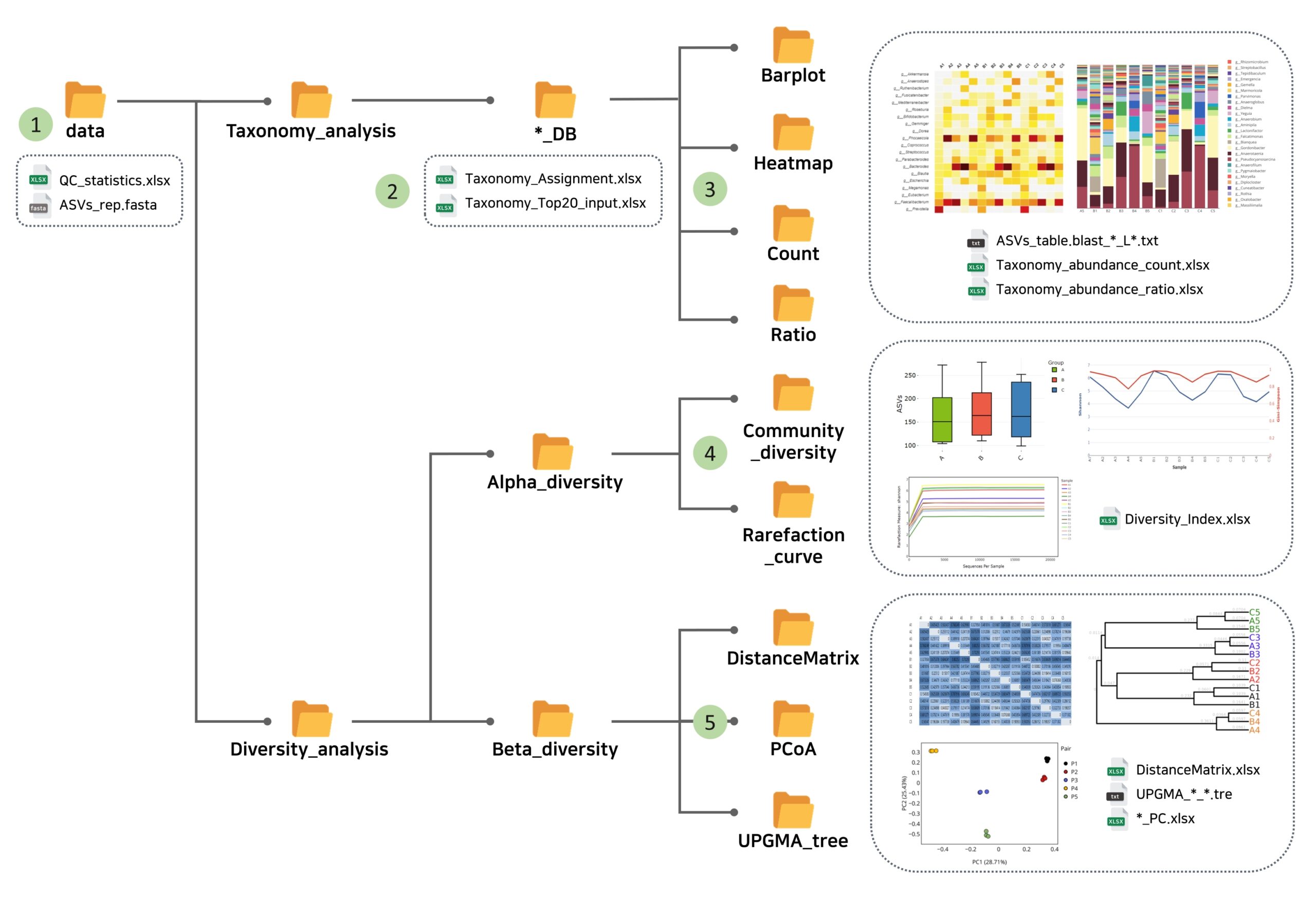
①Summary‐ASV
QC_statistics.xlsx: 生データ、クオリティフィルター、除去されたキメラリードなどの配列数を表したものです。
ASVs_rep.fasta: 各ASVの塩基配列。
② Taxonomy解析(DB)
(_DB)TAXONOMY_Assignment.xlsx:各ASVのTaxonomy解析のエクセルファイル。
Top20_Taxonomy_Input.xlsx: グラフに利用されている上位20のTaxonomyの存在比率を示したファイル。属、種などの各Taxonomyレベルごとにシートを分けて示されています。
③ Taxonomy解析( Count, Ratio)
ASVs_table._L.txt:各Taxonomyレベルごとに微生物の存在比率(または配列数)をまとめたデータを報告します。
Taxonomy_abundance_count.xlsx: 各サンプルに対応するリードカウントを、 Taxonomyレベルごとに別々のシートで表示します。
Taxonomy_abundance_ratio.xlsx: 各サンプルに対する相対的な比率を示しています。
④ Alpha_Diversity (Community_diversity)
Diversity_Index.xlsx: サンプルごとのshannon 、 simpson 、PD_whole_tree、ASV値を1つの表にまとめています。
⑤ Beta_Diversity (DistanceMatrix, PCoA, UPGMA_tree)
_PC.xlsx: PCoA解析の結果。各主成分(PC)の分散と寄与率(%)を示しています。
_DistanceMatrix.xlsx: Bray Curtis, Weighted UniFrac, Unweighted UniFracで定義された各サンプルのDistance Matrixが示されています。
UPGMA_ _.tree: Distance Matrix (Bray Curtis, Weighted UniFrac, Unweighted UniFrac)のUPGMA法によるクラスタリング結果が.tree形式で示されています。
ANALYSIS PROCEDURE
解析全体の流れを記載しています。
納品HTMLの「METHODS」をクリックすると、解析の詳細をまとめた内容を確認できます。
あわせてご確認ください。
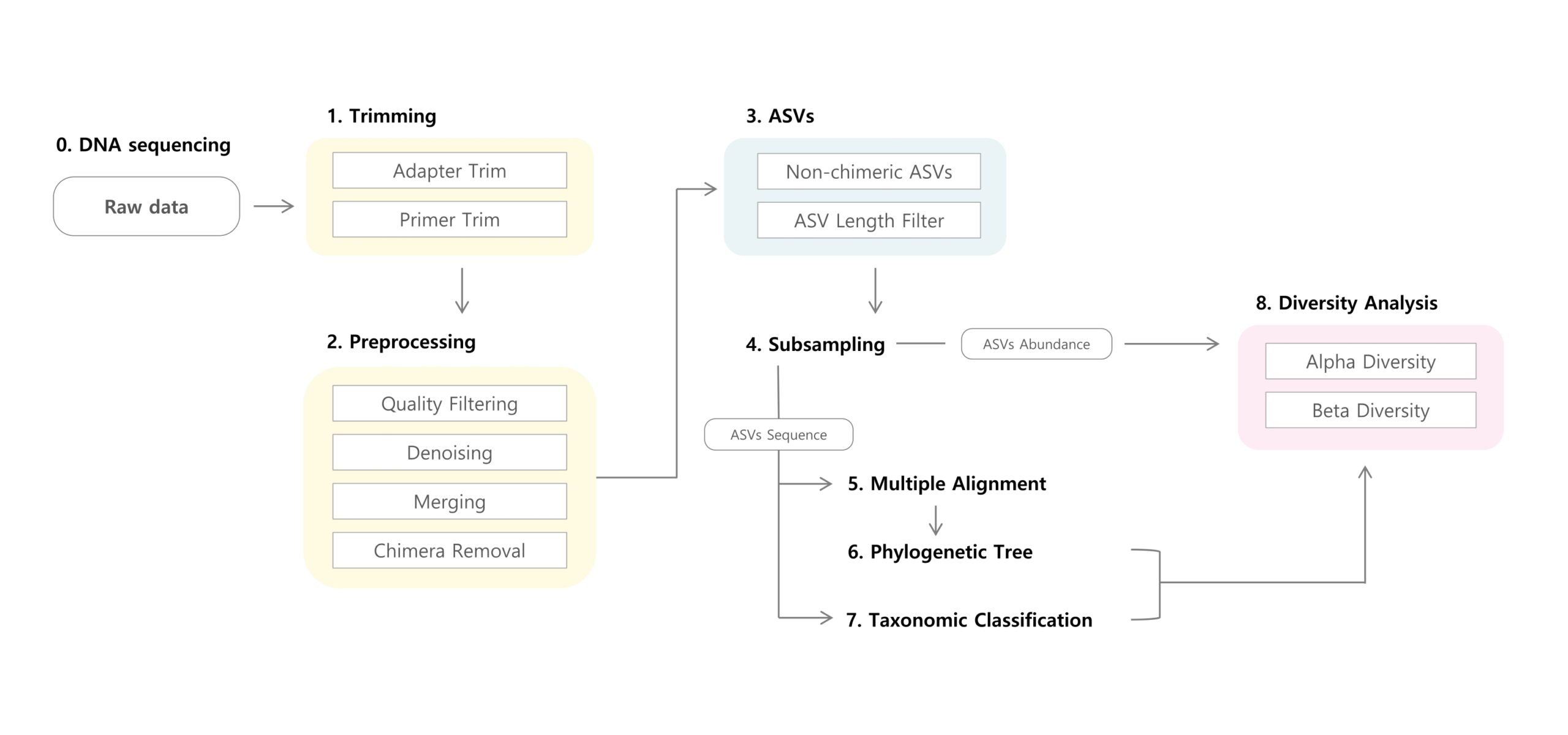
RESULTS OF ANALYSIS
- Rawdata: 生のリードカウント
- Adapter & Primer Trimmingデータ: アダプター/プライマートリミング後のリードカウント
- Preprocessing Length Trimming : 前処理でのLengthトリミング後のリードカウント
- Quality filter : Q値でフィルタリングした後のリードカウント
- denoisedFor: DADA2エラーモデルでフィルターした後の順方向リードカウント
- denoisedRev: DADA2エラーモデルを用いてフィルタリングした後の逆方向のリードカウント
- mergedPair: DADA2 の mergepairs 関数を用いて算出したリードカウント
- non chimeric: キメラを除去した後のリードカウント
- ASV Length Filter: Lengthトリミング後の最終リードカウント
- ASVs:ASVの検出数
- Shannon:種の豊富度と均等度を表す指数
(希少な種の影響を受けやすい) - Simpson:種の豊富度と均等度を表す指数
(主要な種の影響を受けやすい) - 各サンプルの系統学的多様性(PD: phylogenetic diversity)
- graph作成に用いられた値
Bray Curtis
群集が似ているときに 0 となり、全く異なる際に最大値 1 を示します。Weighted UniFrac
微生物の存在量を考慮 (リード数の重みをつけて評価) Unweighted UniFrac
微生物の存在量は考慮せず在、不在で評価- Cutadapt
- DADA2
- QIIME
- Mafft
- FastTreeMP
- BLAST
- Bayesian
- VSEARCH
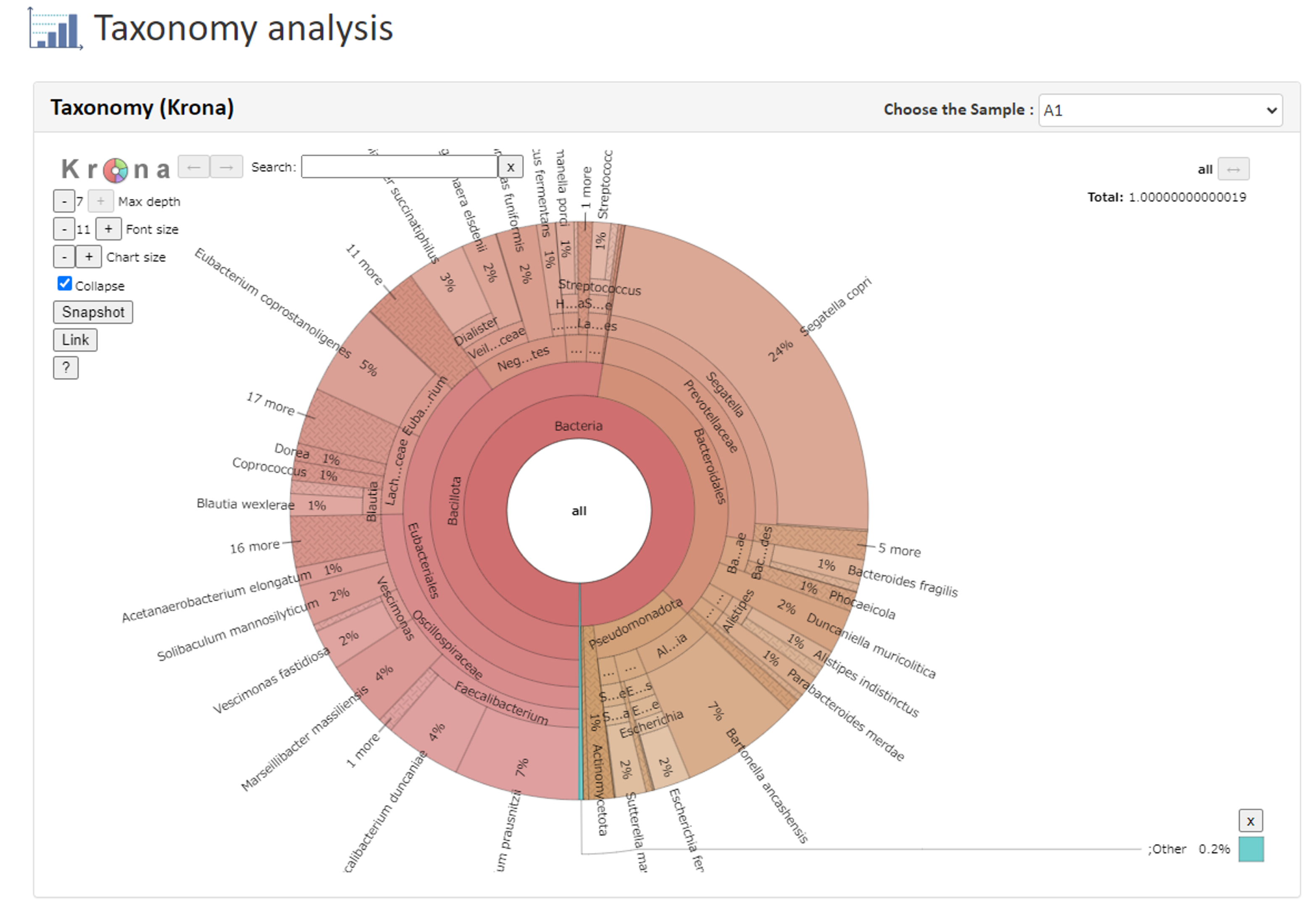
 ②Taxonomy AnalysisTaxonomy Krona
②Taxonomy AnalysisTaxonomy Kronaサンプル毎の群集組成を円グラフで示しています。 グラフの表示名を選択することで、その分類群が占める割合(%)が表示されます。 左上の項目で使いやすいサイズに変更可能です
Taxonomy Bar plot(phylum)各サンプルの分類学的構成を門レベルから属レベルまで示しています。 プロットにマウスオーバーすると分類群がどの割合に該当しているかを確認でき、 上の項目より確認したい分類レベルや使用したい色、グラフに必要な分類を選択可能です。
【分類学的構成(属レベル)を棒グラフで表示した例】
x 軸:サンプル名 y軸: ASV の割合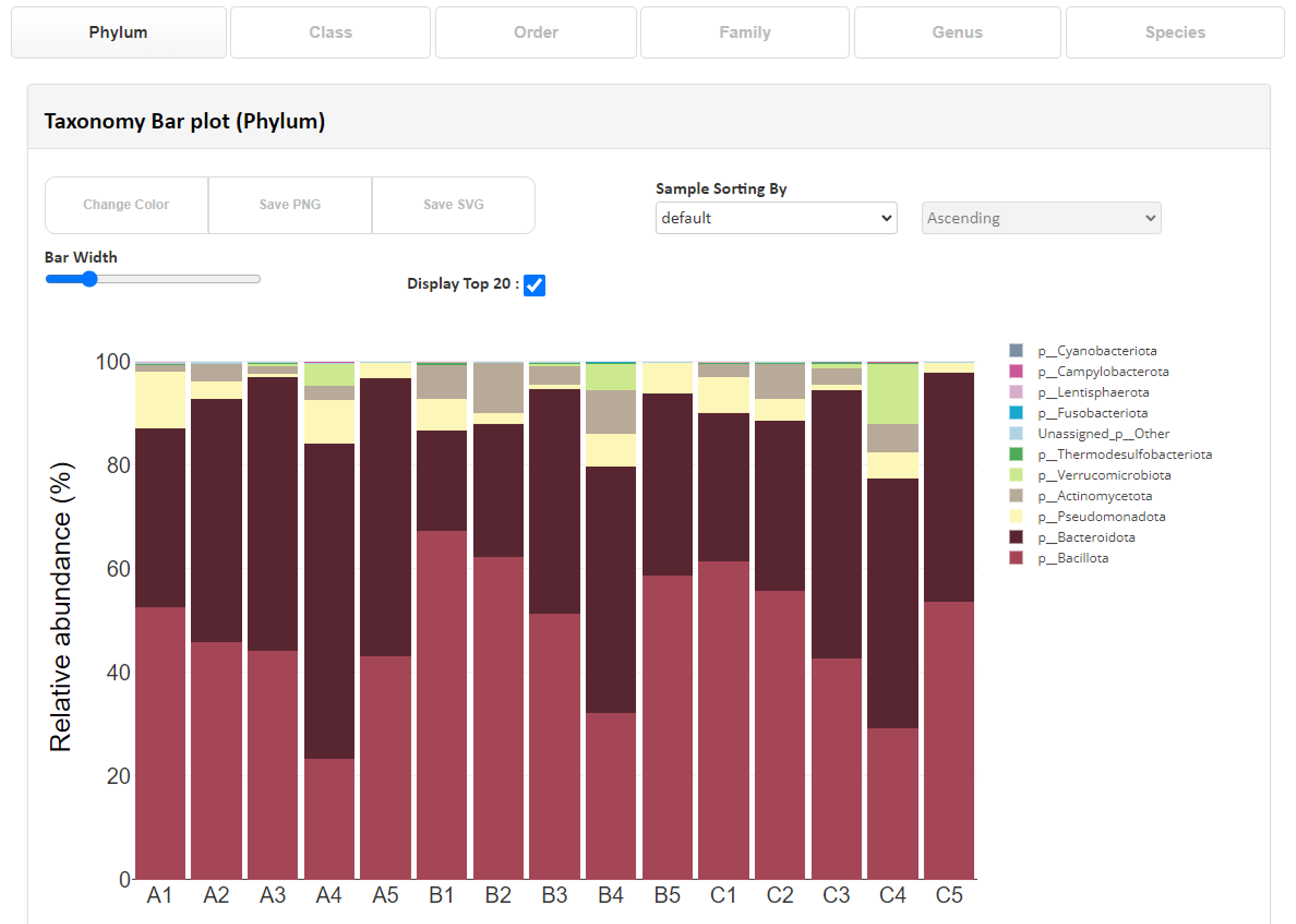 Taxonomy Heatmap polt(phylum)
Taxonomy Heatmap polt(phylum)各サンプルの門から属レベルまでの分類学的構成を示しています。
プロットにマウスオーバーすると、どの分類群が表示された割合に該当しているかを確認できます。
上の項目より分類レベルとカラーパターン等の表示を変更可能です。
(x軸:サンプル名、y軸:上位分類群(最大20)の相対的割合)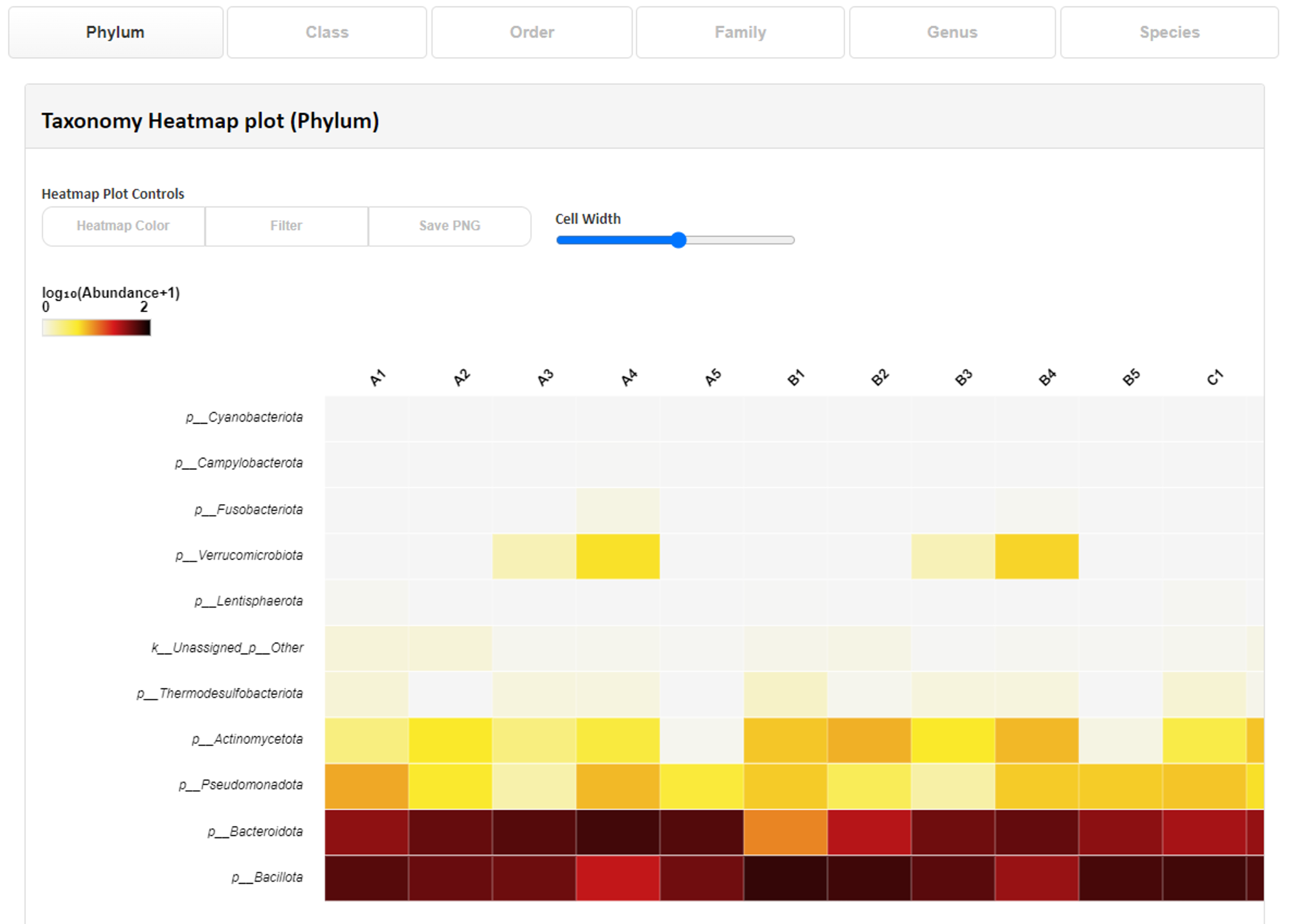 ③ Diversity anaalysisAlpha diversity
③ Diversity anaalysisAlpha diversityサンプル毎のα多様性を記載しています。
※α多様性:ある1つのサンプルの多様性
※β多様性:サンプル間の多様性(類似性・非類似性)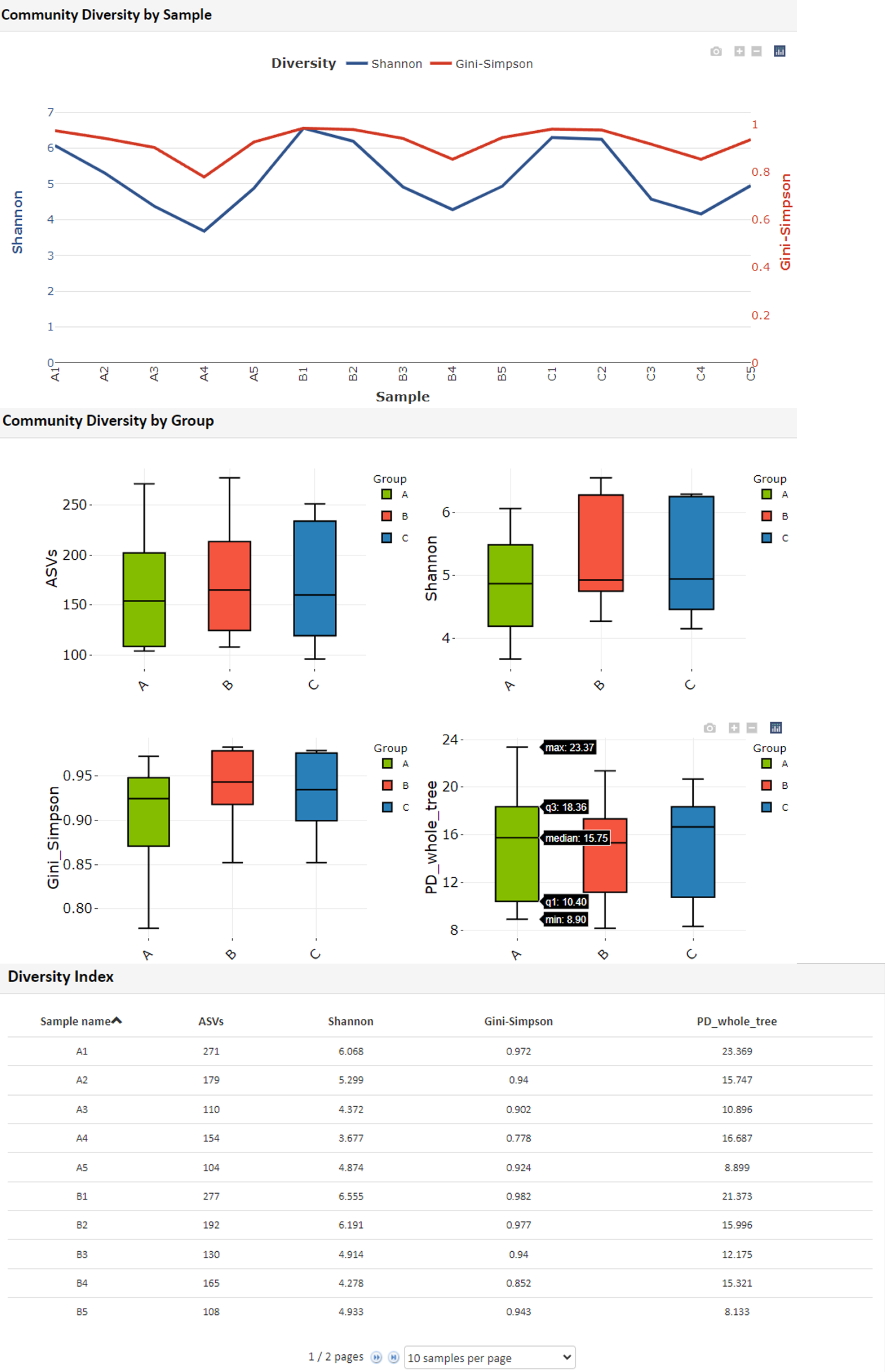
Community Diversity by Sample
・Shannon指数:群集における種の多様性を示す指数。
・Gini-Simpson指数 :群集における種の均等性
(種間の存在量がどの程度似ているか)を示す指数
Community Diversity by Group
Rarefaction Curve(ASVs)rarefaction curve解析に使用されたリード数が種/ASVの同定に十分であったかどうかを示します。 曲線が右に行くほど平坦になる場合は解析に適切な数のリードが使用されたことを示し、追加のシーケンスは必要ありません。 一方、グラフがフラットにならない場合はリードを追加することでそのサンプルについてより多くのASVが発見される可能性があります。
【Rarefaction curveの例】
x軸:各リード数 y軸: ASVs(ASVs, Shannon, Gini-Simpson, PD_Whole_treeで作成)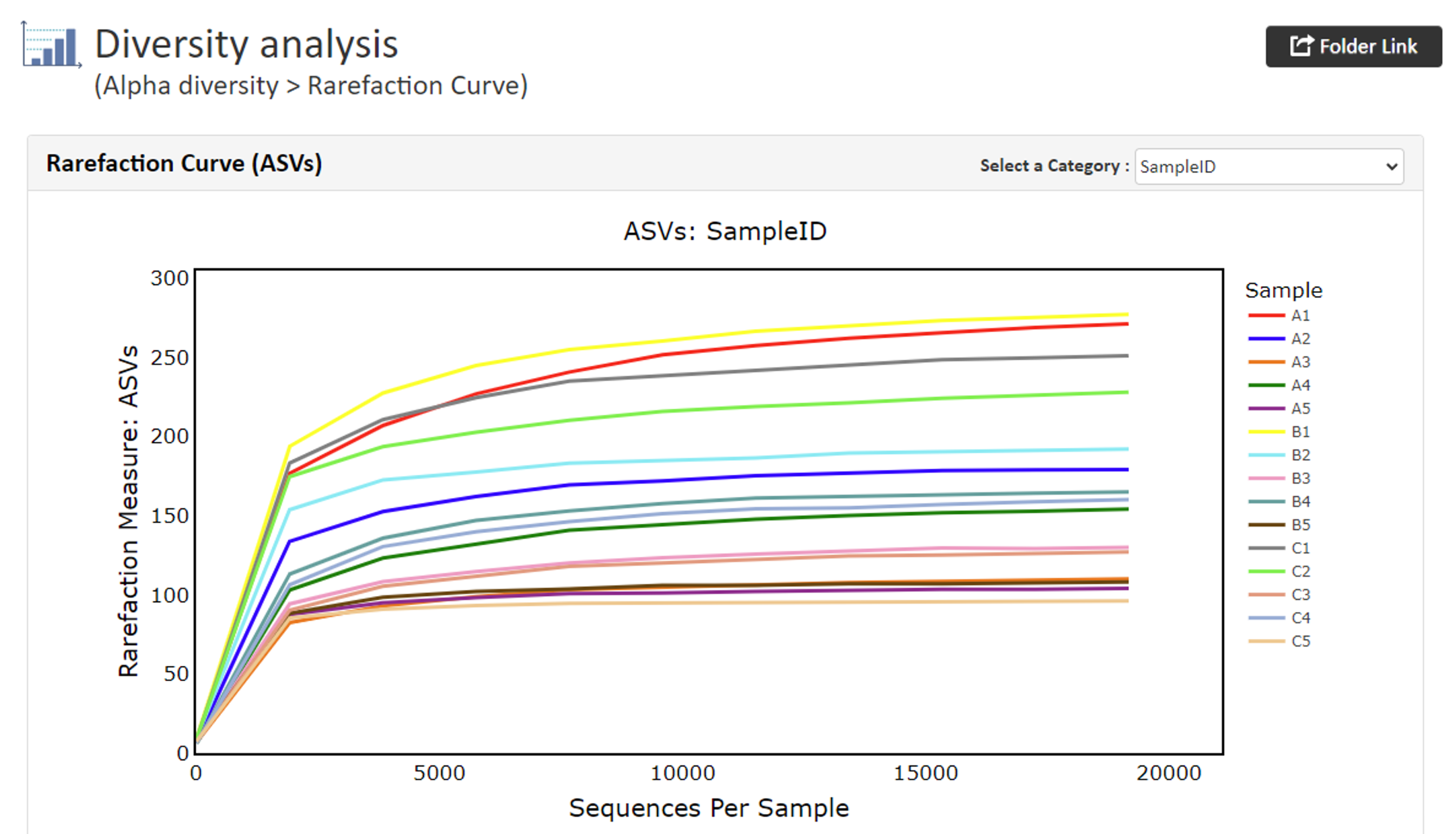 Dastance Matrix(Heatmap)
Dastance Matrix(Heatmap)
【Dastance Matrix Bray Curtisの例】
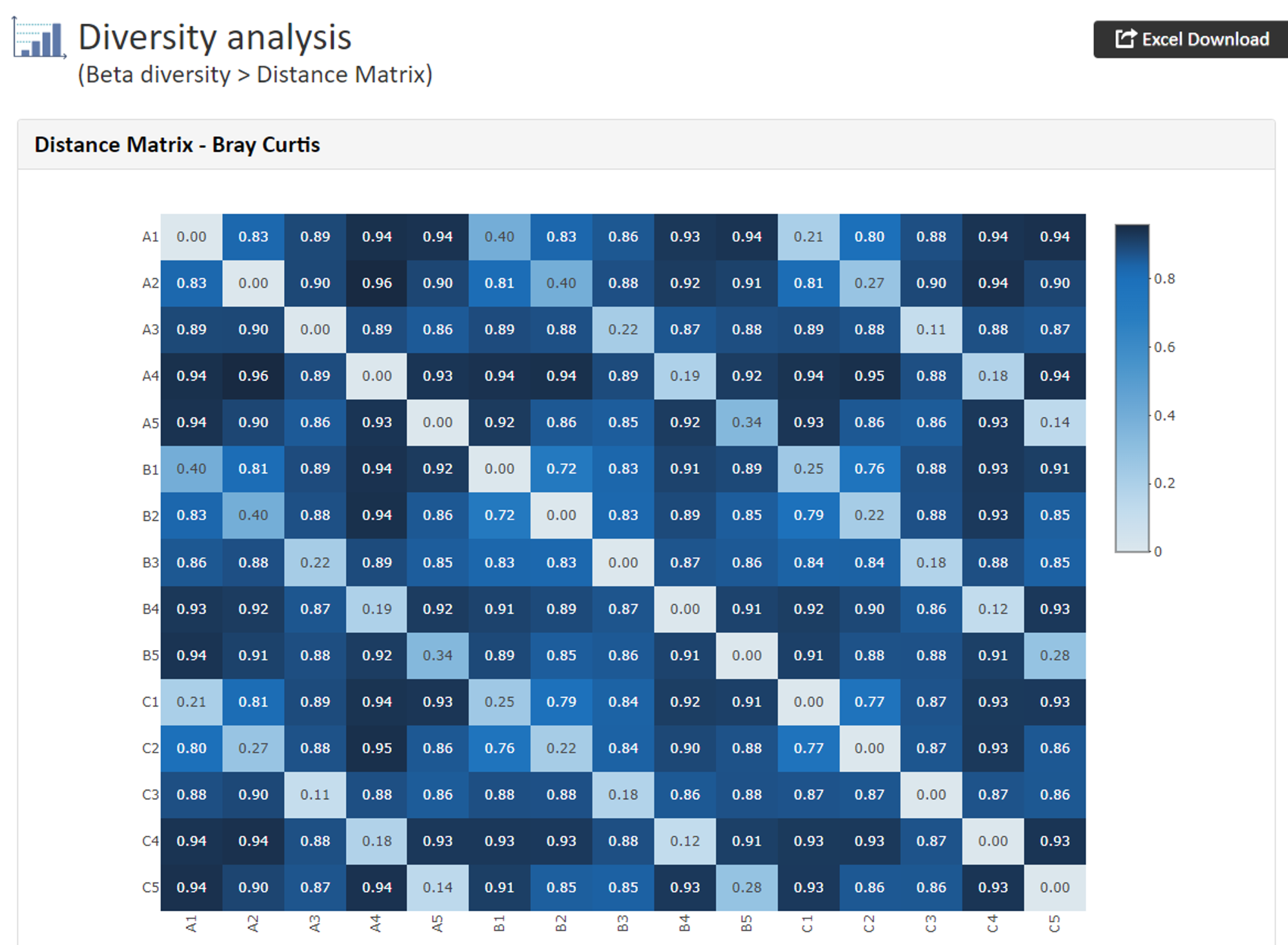 UPGMA tree (平均距離法)
UPGMA tree (平均距離法)サンプル間の類似度(非類似度)を視覚化するために、階層的クラスタリング解析を行った結果です。群集構造が近いサンプル同士でクラスタリングされます。
【UPGMAの結果例】
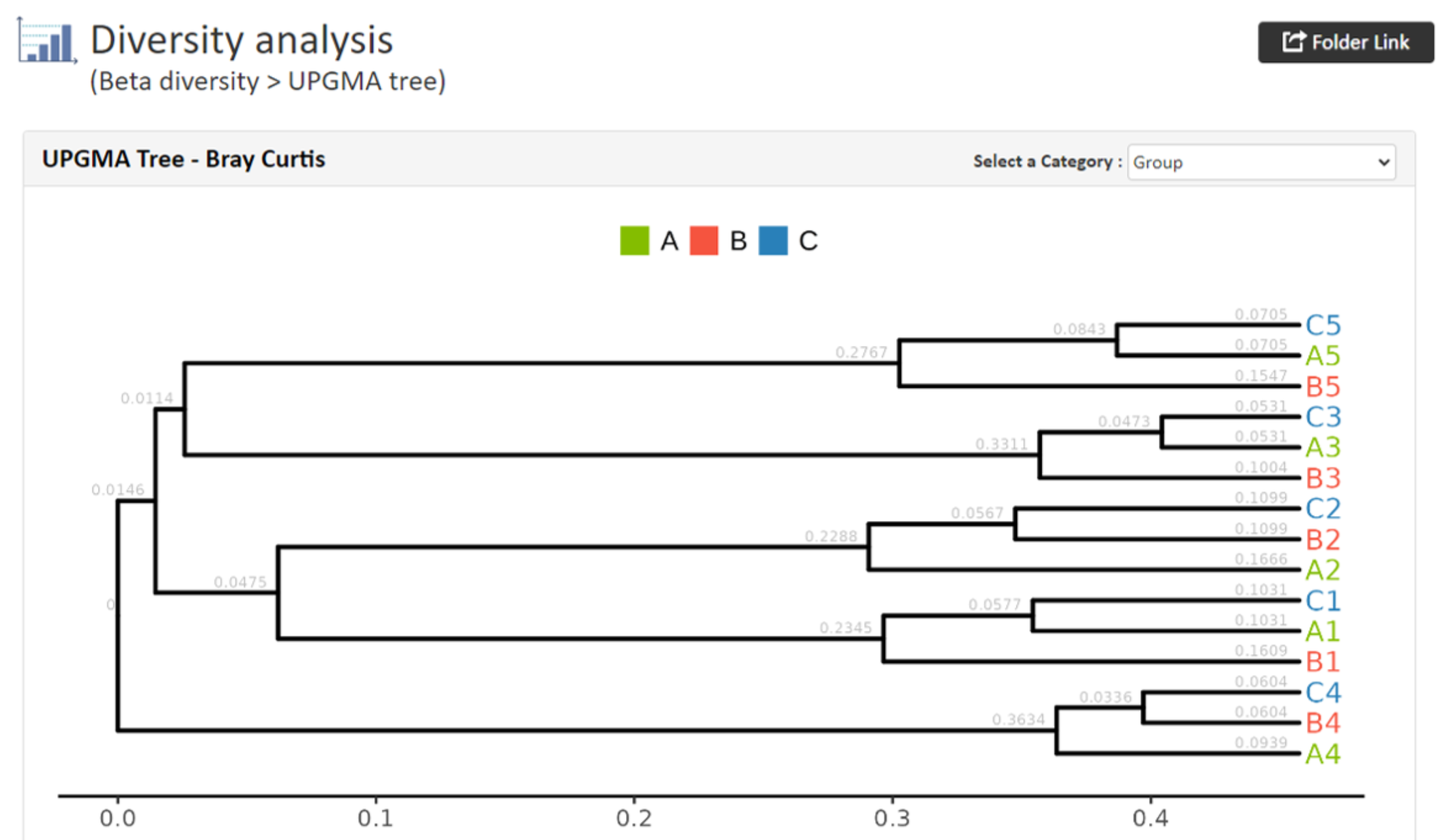 PCoA(主座標分析)
PCoA(主座標分析)PCoAはサンプル間の類似度(非類似度)を視覚化するものです。群集構造が類似しているサンプル同士は近くに、そうでないものは離れてプロットされます。2D、3D(一定のサンプル数が必要) の2種類を作成しております。
【PCoAの結果例(2D)】
各軸には寄与率が与えられ、情報量の何%をその軸で説明できるかを表します。例ではサンプル間の群集構造の違いを数値化したUnifrac distanceを解析に使用しています。
グループ情報を指定した場合、グループごとに色分されたプロットが出力されます。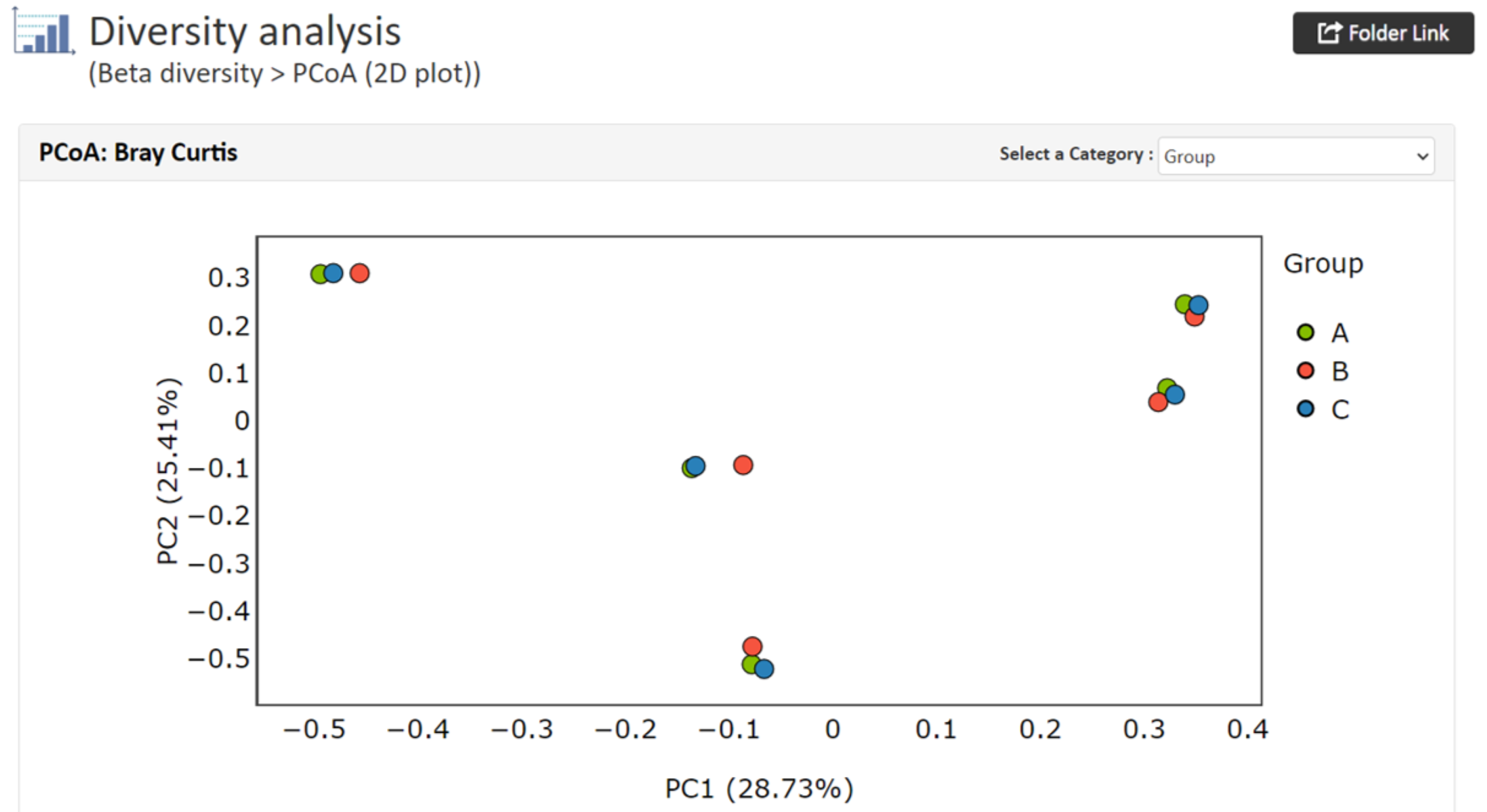
【PCoAの結果例(3D)※サンプル数4サンプルから】
各軸には寄与率が与えられ、情報量の何%をその軸で説明できるかを表します。
例ではサンプル間の群集構造の違いをそれぞれ数値化したデータを使用しています。
グループ情報を指定した場合、グループごとに色分されたプロットが出力されます。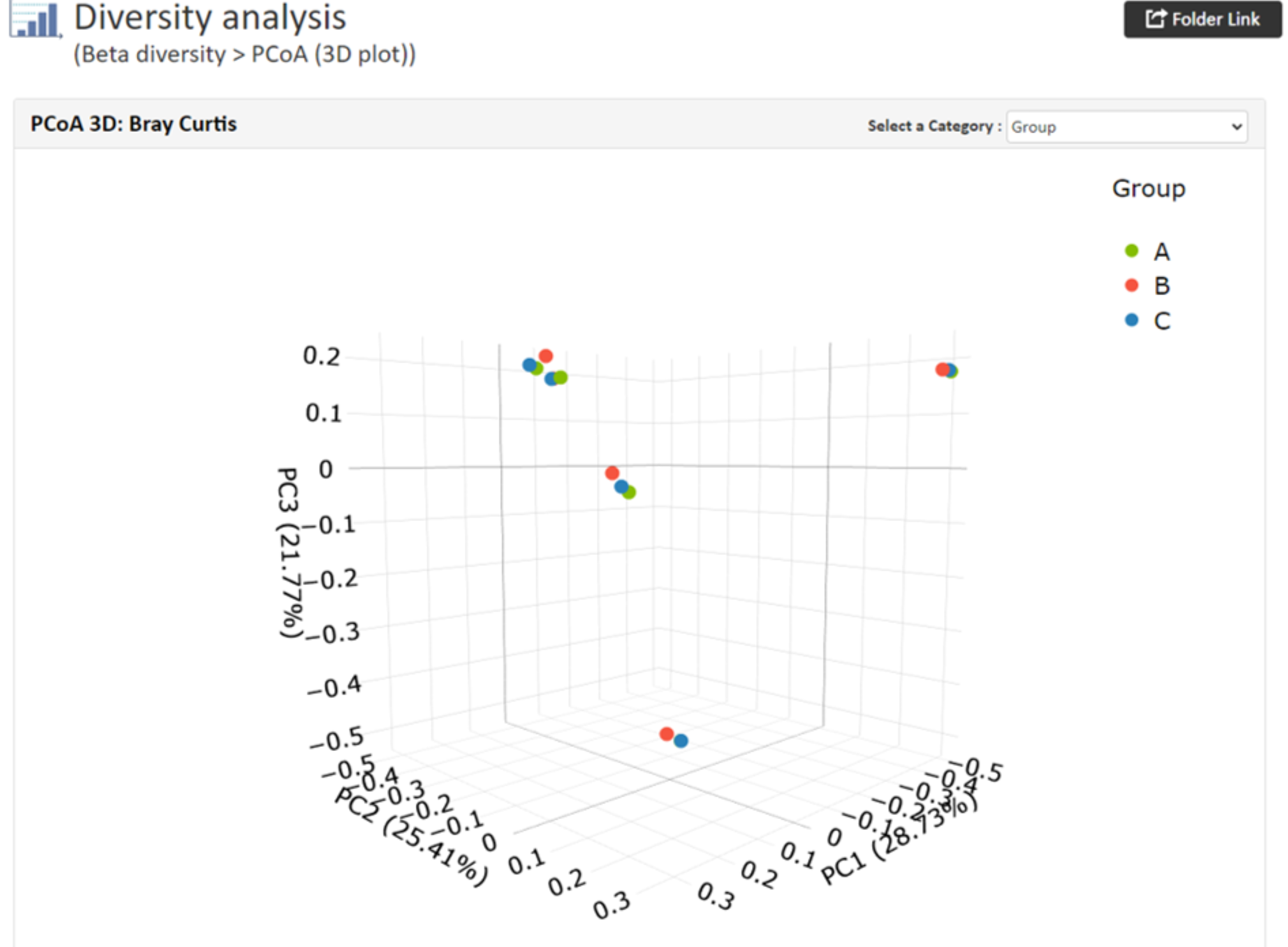
PROGRAM
ASV解析に使用したプログラムCAUTION納品物につきまして「Raw_data 」フォルダ内の report.html が Raw Data に関する報告書です。
報告書の左側のメニューより、リードクオリティの確認、Raw Data(fastqファイル)のダウンロードが可能です。
ASV解析結果は「受託番号 _ASV」フォルダからご確認ください。
Raw data(fastqファイル)は論文投稿時に行うデータベース登録時に、
また、弊社でのデータ保管期間経過後に、追加データ解析をご希望の場合に必要なファイルです。
ダウンロードできる期間は約2週間となっておりますので、必ず期間中に全てのファイルをダウンロードして下さい。
なお、2 週間経過後も1ヵ月はデータを保管しておりますので、再度ダウンロードが必要な場合はngs@macrogen-japan.co.jp までご連絡ください。md5sum値の確認ダウンロードしたFastq.gzファイルもしくはHDD内のFastq.gzファイルは、ファイルの解凍前に”md5sum値”のご確認くだい。
”QuickHash-GUI”という、フリーのアプリケーションもございます。https://www.quickhash-gui.org/downloads/
もしお手持ちのソフトウェアが無ければこちらをご取得下さい。
QuickHash-GUIでのmd5sum値の確認方法は下記となります。
“QuickHash-GUI.exe”アプリケーションを起動します(例1)。
①“FileS”タブをクリックし、
②“Algorithm”、”MD5”を選択してください。md5sum値を確認したいfastqファイルが入っているフォルダを
③“Select Directory” から選択していただきますと、自動的に解析が進行致します。
出力は、csvファイルまたはtxtファイルとして保存することができます。
使用するシステムの性能により、処理に時間がかかる場合がございます。表示された数字と、レポートに記載のmd5sum値の一致を確認できましたら作業完了となります。あわせてQuickHash-GUIのユーザーマニュアルもご確認ください(例2)。例1 QuickHash-GUI起動画面
例2 QuickHash-GUIfile中身
